
The RIESLING (Rapid Identification of EnhancerS LInked to Nearby Genes) ATAC-seq pipeline is designed to be an efficient set of standalone scripts for quickly analyzing ATAC-seq data.
You may find it particularly useful for identifying and stratifying super-enhancers, though can also be leveraged for differential accessibility analysis using DESeq2.
Since this was originally developed in 2014-2015, a number of other packages have been developed, notably the Kundaje lab pipeline and the Ren lab single-cell ATAC pipeline. The Kundaje lab pipeline in particular includes other features (e.g. IDR analysis) which you may find more useful if you aren't interested in super-enhancers / enhancer clusters.
================
- Clone this repo:
git clone https://github.com/GordonLab/riesling-pipeline.git cd riesling-pipeline- Install the Python dependencies:
pip install --user -U -r requirements.txt
================
If you already have a .bed of putative enhancers, you can rapidly derive the super-enhancer population and statistics using get-SuperEnhancers.R. It will not filter blacklisted regions, stitch large regions together, remove TSSes, etc. -- use the full pipeline (detailed below) for that.
A quick example, using the demo-data/sample-mm10-CD4.bed file, which contains signal intensity in the 7th column:
$ git clone https://github.com/GordonLab/riesling-pipeline/
...
$ cd riesling-pipeline/
$ Rscript get-SuperEnhancers.R demo-data/sample-mm10-CD4.bed demo-data/sample-get-SuperEnhancers-output/
[1] "Working on: demo-data/sample-mm10-CD4.bed"
[1] "Output dir: demo-data/sample-get-SuperEnhancers-output/"
[1] "Current directory is: /Users/semenko/git/riesling-pipeline"
[1] "Setting output directory to: demo-data/sample-get-SuperEnhancers-output/"
[1] "Inflection at entry: 24795"
[1] "Corresponding cutoff score: 8105.366159055"
$ cat demo-data/sample-get-SuperEnhancers-output/0-enhancer-stats.txt
Statistics for: demo-data/sample-mm10-CD4.bed
SE Signal %: 38
TE Signal %: 62
SE Count: 1329
TE Count: 24794
SE Count %: 5.09
TE Count %: 94.91
Mean SE Size: 35846.22
Mean TE Size: 5104.87
Median SE Size: 31833
Median TE Size: 892.5
Graphical & .bed results are now in demo-data/sample-get-SuperEnhancers-output/, and will include these figures and more:
| Super-enhancer Cutoff Hockeystick | Super-enhancer Size Distribution | Super vs Traditional vs Stretch Enhancers |
 |
 |
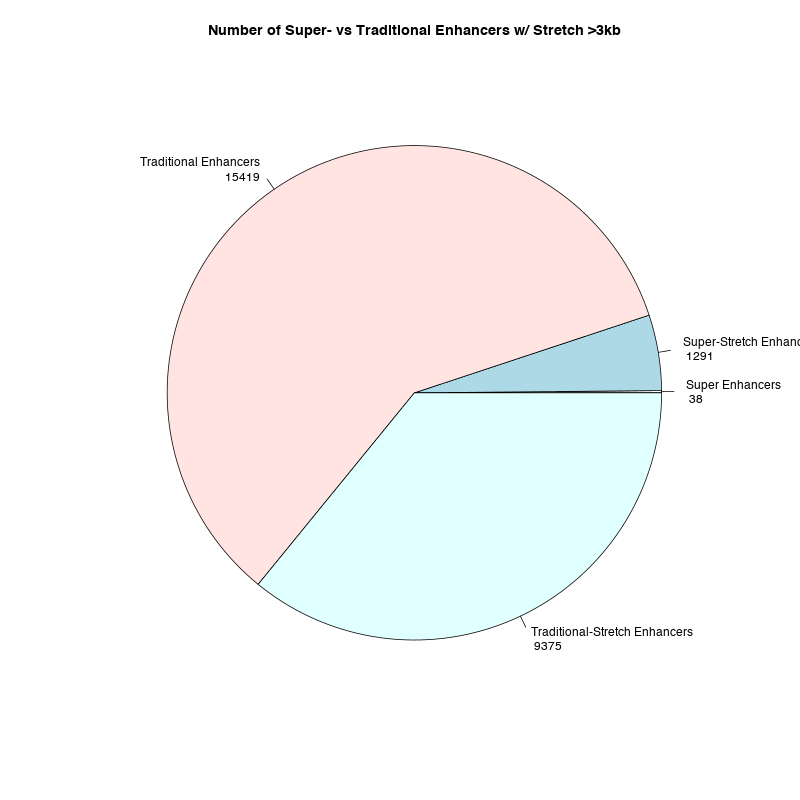 |
Again, this may not be appropriate on non-preprocessed (blacklisted, TSS-filtered, etc.) data. You more likely want to use the full pipeline, detailed below.
The heart of RIESLING and related code expects:
- A .bam file of your aligned ATAC-seq data (e.g. from bowtie2)
- A .bed of peaks (from MACS, HOMER, or any other standard peak caller)
There are lots of ways to pre-process data -- please use whatever approach you prefer. A basic set of standalone scripts to help pre-process paired-end sequencing data is provided, detailed below under 'Pre-processing Tools'.
There are lots of valid way to pre-process data. These standalone scripts may be of use, but please pay careful attention to the peak calling settings you use -- as the defaults may not be applicable to your experimental approach.
-
./1-map-to-genome.py: Run bowtie2 across a folder of paired-end sequence data.
-
./2-sanitize-bam.py: Clean up .bam files, including ATAC-specific fixes. This includes: chrM removal, quality filtering, ENCODE blacklist removal, and PCR duplicate removal
-
./3-call-peaks.py: Run both macs14 and macs2 on .bam files.
By default, this runs both macs14 and macs2, and operates on directories of .bam files.
================