Examples
You can find example-scripts here: MetaMLST Examples. Unpack the archive and follow the instructions below to run MetaMLST on these test HMP samples. In each example, replace the .fastq file with your raw reads to run MetaMLST on your samples.
Be sure to have metamlst scripts folder in your system path:
PATH=<METAMLST_FOLDER_PATH>:$PATH;
Run the following test script: ./metamlst_examples/1_single_sample/test.sh.
The sample FASTQ file SRS013261_epidermidis.fastq contains a subsets of the HMP sample SRS013261. The script executes the following commands:
#Generate a Bowtie2 index from the pre-made database
metamlst-index.py -i bowtie_MmetaMLST
#Map the fastq with Bowtie
bowtie2 --threads 4 --very-sensitive-local -a --no-unal -x bowtie_MmetaMLST -U SRS013261_epidermidis.fastq | samtools view -bS - > SRS013261_epidermidis.bam;
#Run MetaMLST on a single sample
metamlst.py SRS013261_epidermidis.bam -o ./out/
#Type the STs
metamlst-merge.py ./out/
This is a list of the output files produced by MetaMLST at the end of the test-script:
| - | File (in /examples/single_sample) | Type | Description |
|---|---|---|---|
| 1 | sepidermidis.db | MetaMLST Database | Contains STs and sequences |
| 2 | ./out/merged/sepidermidis_report.txt | MetaMLST Report File | Contains the aggregate analysis for all the samples, regarding S. epidermidis. This file contains |
| 3 | ./out/merged/sepidermidis_ST.txt | MetaMLST ST File | Contains the new S. epidermidis ST table after the analys (all the known profiles plus the new profiles detected in the samples). |
Run the following test script: ./metamlst_examples/2_single_sample_custom_db/test.sh.
As for example 1, SRS013261_epidermidis.fastq contains a subsets of the HMP sample SRS013261. The script executes the following commands:
#Create Database with the sequences from MLST_sepidermidis.fasta"
metamlst-index.py -s MLST_sepidermidis.fasta sepidermidis.db
#Create Database with the typings from MLST_sepidermidis_types.txt"
metamlst-index.py -t MLST_sepidermidis_types.txt sepidermidis.db
#Generate a Bowtie2 index
metamlst-index.py -i bowtie_sepidermidis sepidermidis.db
#Map the fastq with Bowtie
bowtie2 --threads 4 --very-sensitive-local -a --no-unal -x bowtie_sepidermidis -U SRS013261_epidermidis.fastq | samtools view -bS - > SRS013261_epidermidis.bam;
#Run MetaMLST on a single sample
metamlst.py -d sepidermidis.db SRS013261_epidermidis.bam -o ./out/
#Type the STs
metamlst-merge.py -d sepidermidis.db ./out
The files produced at the end of the execution are the same of Example 1
Run the following test script: ./metamlst_examples/3_single_sample_multiple_species/test.sh.
MetaMLST is executed on a single file, with the pre-made database, idenifying S. epidermidis and P. acnes. The script executes the following commands:
#Generate a Bowtie2 index
metamlst-index.py -i bowtie_sepidermidis
#Map the fastq with Bowtie
bowtie2 --threads 4 --very-sensitive-local -a --no-unal -x bowtie_sepidermidis -U SRS013261_epidermidis.fastq | samtools view -bS - > SRS013261_epidermidis.bam;
#Run MetaMLST on a single sample
metamlst.py SRS013261_epidermidis.bam -o ./out/
#Type the STs
metamlst-merge.py ./out/
metamlst.py can be run on multiple samples before the MetaMLST-merge step, and can add external metadata to the report files:
Run the following test script: ./metamlst_examples/4_two_samples_with_metadata/test.sh.
#Generate a Bowtie2 index
metamlst-index.py -i bowtie_MmetaMLST
#Map the fastq with Bowtie for each sample
bowtie2 --threads 4 --very-sensitive-local -a --no-unal -x bowtie_MmetaMLST -U SRS015937_epidermidis.fastq | samtools view -bS - > SRS015937_epidermidis.bam;
bowtie2 --threads 4 --very-sensitive-local -a --no-unal -x bowtie_MmetaMLST -U SRS013261_epidermidis.fastq | samtools view -bS - > SRS013261_epidermidis.bam;
#Run MetaMLST on a each sample
metamlst.py SRS015937_epidermidis.bam -o ./out/
metamlst.py SRS013261_epidermidis.bam -o ./out/
#Type the STs using the metadata:
metamlst-merge.py --meta test_metadata.txt ./out/
The script will pair the metadata of the given file with the report-file generated for each species in ./out/merged. The metadata file is a tab-separated table where each row is a sample and each column is a metadata field. The first row is a header. The sampleID (i.e. the name of the file, without extension) must be specified in the first column. A different column can be used, providing the --idField option to metamlst-merge.py. See the related page: metaMLST-merge
MetaMLST output files can be used to generate Phylogenetic Trees based on the reconstructed MLST loci, as well as Minimum Spanning Trees (using tools like PHYLOViZ). In this example, 27 metagenomic files from the HMP (only P. acnes aligning reads are provided) are analyzed with MetaMLST.
Run the following test script: ./metamlst_examples/5_phylogenetic_analysis/test.sh. The script executes the steps of Example 1 on 27 samples, and then executes:
#Type the STs
metamlst-merge.py -d --meta sample_metadata.txt --outseqformat A ./out
Minimum Spanning Trees are a common way to analyse MLST data. Using the typing table (./out/merged/pacnes_ST.txt) and the report file (./out/merged/pacnes_report.txt) you can generate a Minimum Spanning Tree with PHYLOViZ) (the report file can be used as an isolate file to colour the graph according to the metadata):
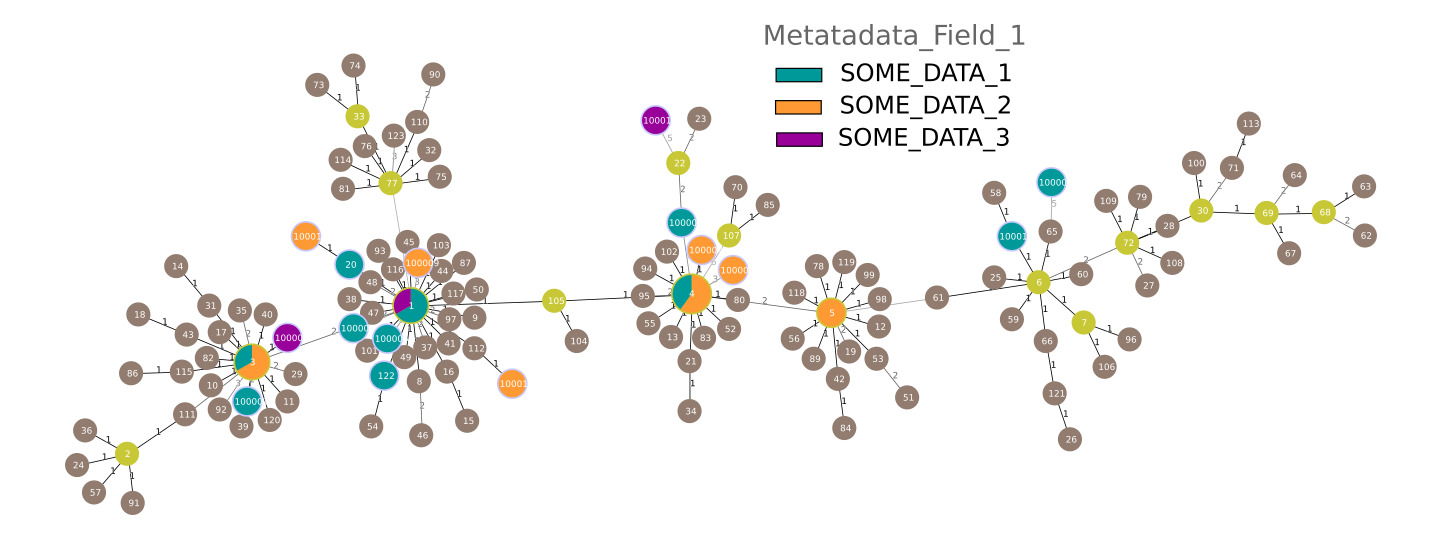
▲ A Minimum Spanning Tree generated from the 27 genomes coloured by metadata field "Metadata_Field_1) plus the available Reference STs for P. acnes (brown)
Using the --outseqformat A option in MetaMLST-merge, you can generate an additional file: ./out/merged/pacnes_sequences.fna, containing the aligned and concatenated sequences of each locus of the 27 samples analysed. By default (see metamlst-merge.py) there is one entry for each sample.
This file can be supplied directly to any phylogenetic-tree software such as RAxML and the tree can be viewd with Archaeopteryx:
mkdir ~/5_phylogenetic_analysis_trees/
raxmlHPC-PTHREADS-SSE3 -T 4 -m GTRCAT -s ./out/merged/pacnes_sequences.fna -w ~/5_phylogenetic_analysis_trees/ -n pacnes_trees -p 12345;
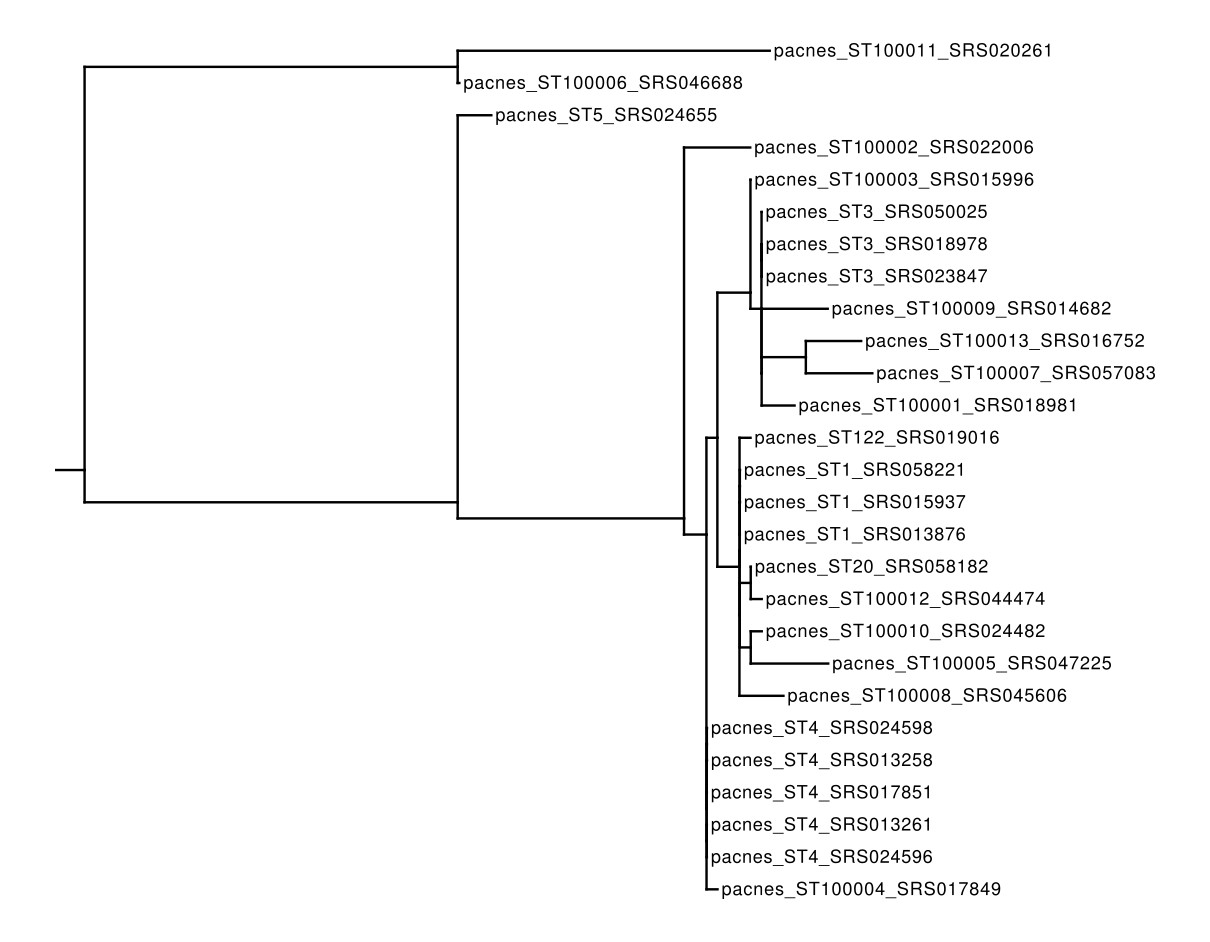
▲ A Phylogenetic Tree built with RAxML on the concatenated MLST loci of the 27 samples analysed in this example
MetaMLST allows to update the database with newly detected sequnces and typing. This can be useful in case of re-detection of a new ST while analysing a different sample, or to cross-compare different dataset analysed in different times.
Run the following test script: ./metamlst_examples/6_reuse_the_db/test.sh. The script executes the steps of Example 4 on the SRS015937 sample, that harbors a new ST of S. epidermidis. The script then updates the database with:
- The updated sequences (./out/merged/sepidermidis_sequences.fna) (generated with the
--outseqformat Bof MetaMLST-merge) - The updated ST table (./out/merged/sepidermidis_ST.txt), modified to include "#sepidermidis|Staphylococcus epidermidis" as first line.
The pipeline is then re-run on the same sample, but with the new Database. The ST-100001 is then considered as "Known" (i.e. previously detected)
See the test script for furhter details.