This Tutorial demonstrates how to use Sunny's histogram binning capabilities (via intensities_binned). This functionality allows the simulation data produced by Sunny to be compared to experimental data produced by Inelastic Neutron Scattering (INS) in an apples-to-apples fashion. Experimental data can be loaded from a MDHistoWorkspace stored in a .nxs file by the Mantid software using load_nxs.
For this example, we will examine the CTFD compound, which is crystallographically approximately a square lattice. We specify the crystal lattice structure of CTFD using the lattice parameters specified by
W Wan et al 2020 J. Phys.: Condens. Matter 32 374007 DOI 10.1088/1361-648X/ab757a
This Tutorial demonstrates how to use Sunny's histogram binning capabilities (via intensities_binned). This functionality allows the simulation data produced by Sunny to be compared to experimental data produced by Inelastic Neutron Scattering (INS) in an apples-to-apples fashion. Experimental data can be loaded from a MDHistoWorkspace stored in a .nxs file by the Mantid software using load_nxs.
For this example, we will examine the CTFD compound, which is crystallographically approximately a square lattice. We specify the crystal lattice structure of CTFD using the lattice parameters specified by
W Wan et al 2020 J. Phys.: Condens. Matter 32 374007 DOI 10.1088/1361-648X/ab757a
latvecs = lattice_vectors(8.113,8.119,12.45,90,100,90)
positions = [[0,0,0]]
types = ["Cu"]
formfactors = [FormFactor(1,"Cu2")]
@@ -15,21 +15,15 @@
for i in 1:10_000 # Long enough to reach equilibrium
step!(sys, langevin)
end
The neutron spectrometer used in the experiment had an incident neutron energy of 36.25 meV. Since this is the most amount of energy that can be deposited by the neutron into the sample, we don't need to consider energies higher than this.
ωmax = 36.25;
We choose the resolution in energy (specified by the number of nω modes resolved) to be ≈20× better than the experimental resolution in order to demonstrate the effect of over-resolving in energy.
We re-sample from the thermal equilibrium distribution several times to increase our sample size
nsamples = 3
for _ in 1:nsamples
for _ in 1:8000
step!(sys, langevin)
end
- add_sample!(dsf, sys)
+ add_sample!(sc, sys)
end
Since the SU(N)NY crystal has only finitely many lattice sites, there are finitely many ways for a neutron to scatter off of the sample. We can visualize this discreteness by plotting each possible Qx and Qz, for example:
# Compute some scattering vectors at and around the first BZ...
-scatter!(ax,Qx,Qz)
One way to display the structure factor is to create a histogram with one bin centered at each discrete scattering possibility using unit_resolution_binning_parameters to create a set of BinningParameters.
params = unit_resolution_binning_parameters(dsf)
Binning Parameters
+scatter!(ax,Qx,Qz)
One way to display the structure factor is to create a histogram with one bin centered at each discrete scattering possibility using unit_resolution_binning_parameters to create a set of BinningParameters.
params = unit_resolution_binning_parameters(sc)
Binning Parameters⊡ 6 bins from -0.083 to +0.917 along [+1.29 dx] (Δ = 0.129)
⊡ 6 bins from -0.083 to +0.917 along [+1.29 dy] (Δ = 0.129)
⊡ 4 bins from -0.125 to +0.875 along [-0.34 dx +1.95 dz] (Δ = 0.126)
@@ -39,23 +33,23 @@
∫ Integrated from -0.083 to +1.750 along [+1.29 dy] (Δ = 1.419)
⊡ 4 bins from -0.125 to +0.875 along [-0.34 dx +1.95 dz] (Δ = 0.126)
∫ Integrated from -0.041 to +77.745 along [+1.00 dE] (Δ = 77.786)
-
Now that we have parameterized the histogram, we can bin our data. In addition to the BinningParameters, an intensity_formula needs to be provided to specify which dipole, temperature, and atomic form factor corrections should be applied during the intensity calculation.
Now that we have parameterized the histogram, we can bin our data. In addition to the BinningParameters, an intensity_formula needs to be provided to specify which dipole, temperature, and atomic form factor corrections should be applied during the intensity calculation.
Let's make a new histogram which includes the energy axis. The x-axis of the histogram will be a 1D cut from Q = [0,0,0] to Q = [1,1,0]. See slice_2D_binning_parameters.
Let's make a new histogram which includes the energy axis. The x-axis of the histogram will be a 1D cut from Q = [0,0,0] to Q = [1,1,0]. See slice_2D_binning_parameters.
Binning Parameters⊡ 10 bins from -0.079 to +1.493 along [+0.91 dx +0.91 dy] (Δ = 0.122)
∫ Integrated from -0.150 to +0.150 along [-0.91 dx +0.91 dy] (Δ = 0.232)
∫ Integrated from -0.150 to +0.150 along [+0.34 dx -1.95 dz] (Δ = 0.151)
⊡ 480 bins from -0.041 to +38.893 along [+1.00 dE] (Δ = 0.081)
-
There are no longer any scattering vectors exactly in the plane of the cut. Instead, as described in the BinningParameters output above, the transverse Q directions are integrated over, so slightly out of plane points are included.
We plot the intensity on a log-scale to improve visibility.
There are no longer any scattering vectors exactly in the plane of the cut. Instead, as described in the BinningParameters output above, the transverse Q directions are integrated over, so slightly out of plane points are included.
We plot the intensity on a log-scale to improve visibility.
If we had used a slower annealing procedure, involving 100,000 or more Langevin time-steps, it would very likely find the correct ground state. Instead, for purposes of illustration, let's analyze the imperfect spin configuration currently stored in sys.
An experimental probe of magnetic order order is the 'instantaneous' or 'static' structure factor intensity, available via InstantStructureFactor and related functions. To infer periodicities of the magnetic supercell, however, it is sufficient to look at the structure factor weights of spin sublattices individually, without phase averaging. This information is provided by print_wrapped_intensities (see the API documentation for a physical interpretation).
print_wrapped_intensities(sys)
Dominant wavevectors for spin sublattices:
+end
Because the quench was relatively fast, it is expected to find defects in the magnetic order. These can be visualized.
If we had used a slower annealing procedure, involving 100,000 or more Langevin time-steps, it would very likely find the correct ground state. Instead, for purposes of illustration, let's analyze the imperfect spin configuration currently stored in sys.
An experimental probe of magnetic order order is the 'instantaneous' or 'static' structure factor intensity, available via instant_correlations and related functions. To infer periodicities of the magnetic supercell, however, it is sufficient to look at the structure factor weights of spin sublattices individually, without phase averaging. This information is provided by print_wrapped_intensities (see the API documentation for a physical interpretation).
print_wrapped_intensities(sys)
Dominant wavevectors for spin sublattices:
[1/4, 0, 1/4] 49.79% weight
[-1/4, 0, -1/4] 49.79%
@@ -136,7 +136,7 @@
[0, 0, 0]]
labels = ["($(p[1]),$(p[2]),$(p[3]))" for p in points]
density = 600
-path, markers = connected_path(swt, points, density);
dispersion may now be called on the wave vectors along the generated path. Each column of the returned matrix corresponds to a different mode.
disp = dispersion(swt, path);
In addition to the band energies $\omega_i$, Sunny can calculate the inelastic neutron scattering intensity $I(q,\omega_i(q))$ according to an intensity_formula. The default formula applies a polarization correction $(1 - Q\otimes Q)$.
formula = intensity_formula(swt; kernel = delta_function_kernel)
Quantum Scattering Intensity Formula
+path, markers = connected_path(swt, points, density);
dispersion may now be called on the wave vectors along the generated path. Each column of the returned matrix corresponds to a different mode.
disp = dispersion(swt, path);
In addition to the band energies $\omega_i$, Sunny can calculate the inelastic neutron scattering intensity $I(q,\omega_i(q))$ according to an intensity_formula. The default formula applies a polarization correction $(1 - Q\otimes Q)$.
formula = intensity_formula(swt; kernel = delta_function_kernel)
Quantum Scattering Intensity Formula
At any Q and for each band ωᵢ = εᵢ(Q), with S = S(Q,ωᵢ):
Intensity(Q,ω) = ∑ᵢ δ(ω-ωᵢ) ∑_ij (I - Q⊗Q){i,j} S{i,j}
@@ -174,25 +174,25 @@
)
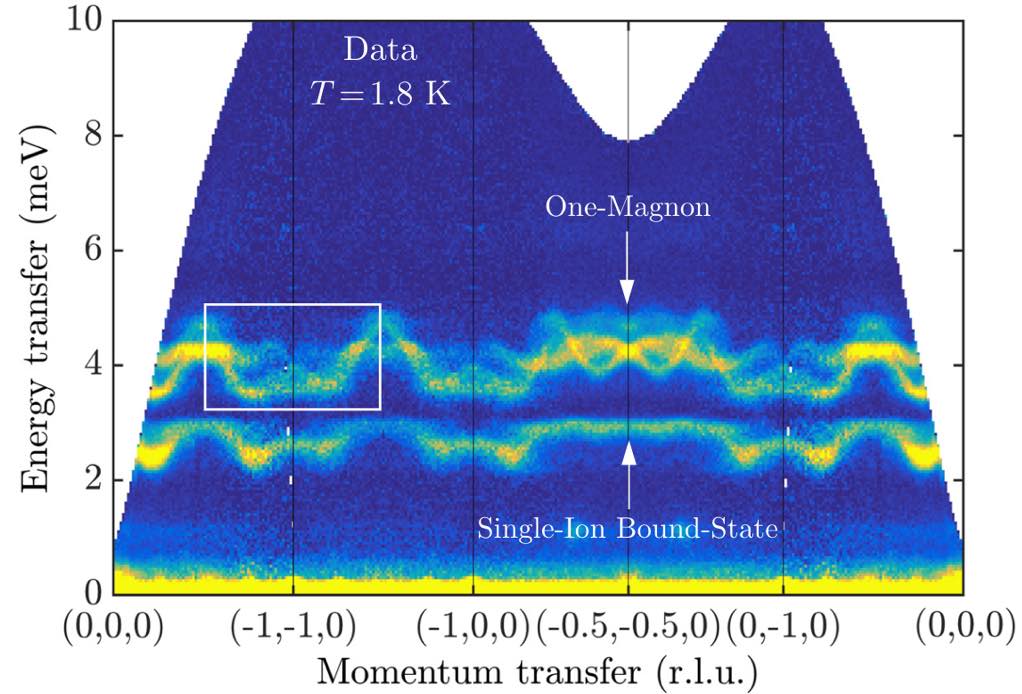
The existence of a lower-energy, single-ion bound state is in qualitative agreement with the experimental data in Bai et al. (Note that the publication figure uses a different coordinate system to label the same wave vectors and the experimental data necessarily averages over the three degenerate ground states.)
The full data from the dynamical spin structure factor (DSSF) can be retrieved with the dssf function. Like dispersion and intensities, dssf takes an array of wave vectors.
disp, Sαβ = dssf(swt, [[0, 0, 0]]);
disp is identical to the output that is obtained from dispersion and contains the energy of each mode at the specified wave vectors. Sαβ contains a 3x3 matrix for each of these modes. The matrix elements of Sαβ correspond to correlations of the different spin components (ordered x, y, z). For example, the full set of matrix elements for the first mode may be obtained as follows,
Sαβ[1]
3×3 StaticArraysCore.SMatrix{3, 3, ComplexF64, 9} with indices SOneTo(3)×SOneTo(3):
3.45531e-30+0.0im … 6.90657e-32+1.76065e-32im
-2.76136e-31-3.60465e-32im -5.3358e-33-2.12755e-33im
- -0.133536+0.105808im 1.47021e-33+0.0im
Linear spin wave calculations are very useful for getting quick, high-quality, results at zero temperature. Moreover, these results are obtained in the thermodynamic limit. Classical dynamics may also be used to produce similar results, albeit at a higher computational cost and on a finite sized lattice. The classical approach nonetheless provides a number of complementary advantages: it is possible perform simulations at finite temperature while retaining nonlinearities; out-of-equilibrium behavior may be examined directly; and it is straightforward to incorporate inhomogenties, chemical or otherwise.
Because classical simulations are conducted on a finite-sized lattice, obtaining acceptable resolution in momentum space requires the use of a larger system size. We can now resize the magnetic supercell to a much larger simulation volume, provided as multiples of the original unit cell.
Linear spin wave calculations are very useful for getting quick, high-quality, results at zero temperature. Moreover, these results are obtained in the thermodynamic limit. Classical dynamics may also be used to produce similar results, albeit at a higher computational cost and on a finite sized lattice. The classical approach nonetheless provides a number of complementary advantages: it is possible perform simulations at finite temperature while retaining nonlinearities; out-of-equilibrium behavior may be examined directly; and it is straightforward to incorporate inhomogenties, chemical or otherwise.
Because classical simulations are conducted on a finite-sized lattice, obtaining acceptable resolution in momentum space requires the use of a larger system size. We can now resize the magnetic supercell to a much larger simulation volume, provided as multiples of the original unit cell.
Apply Langevin dynamics to thermalize the system to a target temperature.
kT = 0.5 * meV_per_K # 0.5K in units of meV
langevin.kT = kT
for _ in 1:10_000
step!(sys_large, langevin)
-end
We can measure the DynamicStructureFactor by integrating the Hamiltonian dynamics of SU(N) coherent states. Three keyword parameters are required to determine the ω information that will be calculated: an integration step size, the number of ωs to resolve, and the maximum ω to resolve. For the time step, twice the value used for the Langevin integrator is usually a good choice.
sf = DynamicStructureFactor(sys_large; Δt=2Δt, nω=120, ωmax=7.5)
To estimate the dynamic structure factor, we can collect spin-spin correlation data by first generating an initial condition at thermal equilibrium and then integrating the Hamiltonian dynamics of SU(N) coherent states. Samples are accumulated into a SampledCorrelations, which is initialized by calling dynamical_correlations. dynamical_correlations takes a System and three keyword parameters that determine the ω information that will be available: an integration step size, the number of ωs to resolve, and the maximum ω to resolve. For the time step, twice the value used for the Langevin integrator is usually a good choice.
sf currently contains dynamical structure data generated from a single sample. Additional samples can be added by generating a new spin configuration and calling add_sample!:
for _ in 1:2
+
sc currently contains no data. A sample can be accumulated into it by calling add_sample!.
add_sample!(sc, sys_large)
Additional samples can be added after generating new spin configurations:
for _ in 1:2
for _ in 1:1000 # Fewer steps needed in equilibrium
step!(sys_large, langevin)
end
- add_sample!(sf, sys_large) # Accumulate the sample into `sf`
-end
The basic functions for accessing intensity data are intensities_interpolated and intensities_binned. Both functions accept an intensity_formula to specify how to combine the correlations recorded in the StructureFactor into intensity data. By default, a formula computing the unpolarized intensity is used, but alternative formulas can be specified.
By way of example, we will use a formula which computes the trace of the structure factor and applies a classical-to-quantum temperature-dependent rescaling kT.
formula = intensity_formula(sf, :trace; kT = kT)
Classical Scattering Intensity Formula
+ add_sample!(sc, sys_large) # Accumulate the sample into `sc`
+end
The basic functions for accessing intensity data are intensities_interpolated and intensities_binned. Both functions accept an intensity_formula to specify how to combine the correlations recorded in the SampledCorrelations into intensity data. By default, a formula computing the unpolarized intensity is used, but alternative formulas can be specified.
By way of example, we will use a formula which computes the trace of the structure factor and applies a classical-to-quantum temperature-dependent rescaling kT.
formula = intensity_formula(sc, :trace; kT = kT)
Classical Scattering Intensity Formula
At discrete scattering modes S = S[ix_q,ix_ω], use:
Intensity[ix_q,ix_ω] = Tr S
@@ -200,13 +200,13 @@
No form factors specifiedTemperature corrected (kT = 0.043086666310725885) ✓
Using the formula, we plot single-$q$ slices at (0,0,0) and (π,π,π):
For real calculations, one often wants to apply further corrections and more accurate formulas Here, we apply FormFactor corrections appropriate for Fe2 magnetic ions, and a dipole polarization correction :perp.
Classical Scattering Intensity Formula
At discrete scattering modes S = S[ix_q,ix_ω], use:
Intensity[ix_q,ix_ω] = ∑_ij (I - Q⊗Q){i,j} S{i,j}
@@ -222,35 +222,35 @@
[0, 1, 0],
[0, 0, 0]]
density = 40
-path, markers = connected_path(sf, points, density);
Since scattering intensities are only available at a certain discrete $(Q,\omega)$ points, the intensity on the path can be calculated by interpolating between these discrete points:
is = intensities_interpolated(sf, path;
+path, markers = connected_path(sc, points, density);
Since scattering intensities are only available at a certain discrete $(Q,\omega)$ points, the intensity on the path can be calculated by interpolating between these discrete points:
is = intensities_interpolated(sc, path;
interpolation = :linear, # Interpolate between available wave vectors
formula = new_formula
)
-is = broaden_energy(sf, is, (ω, ω₀)->lorentzian(ω-ω₀, 0.05)) # Add artificial broadening
+is = broaden_energy(sc, is, (ω, ω₀)->lorentzian(ω-ω₀, 0.05)) # Add artificial broadening
labels = ["($(p[1]),$(p[2]),$(p[3]))" for p in points]
-heatmap(1:size(is,1), ωs(sf), is;
+heatmap(1:size(is,1), ωs(sc), is;
axis = (
ylabel = "meV",
xticks = (markers, labels),
xticklabelrotation=π/8,
xticklabelsize=12,
)
-)
Whereas intensities_interpolated either rounds or linearly interpolates between the discrete $(Q,\omega)$ points Sunny calculates correlations at, intensities_binned performs histogram binning analgous to what is done in experiments. The graphs produced by each method are similar.
cut_width = 0.3
+)
Whereas intensities_interpolated either rounds or linearly interpolates between the discrete $(Q,\omega)$ points Sunny calculates correlations at, intensities_binned performs histogram binning analgous to what is done in experiments. The graphs produced by each method are similar.
Note that Brillouin zones appear 'skewed'. This is a consequence of the fact that Sunny measures $q$-vectors as multiples of reciprocal lattice vectors, which are not orthogonal. It is often useful to express our wave vectors in terms of an orthogonal basis, where each basis element is specified as a linear combination of reciprocal lattice vectors. For our crystal, with reciprocal vectors $a^*$, $b^*$ and $c^*$, we can define an orthogonal basis by taking $\hat{a}^* = 0.5(a^* + b^*)$, $\hat{b}^*=a^* - b^*$, and $\hat{c}^*=c^*$. Below, we map qs to wavevectors ks in the new coordinate system and get their intensities.
Finally, we note that instantaneous structure factor data, $𝒮(𝐪)$, can be obtained from a dynamic structure factor with instant_intensities_interpolated.
Finally, we note that instantaneous structure factor data, $𝒮(𝐪)$, can be obtained from a dynamic structure factor with instant_intensities_interpolated.