
This result can be directly compared to experimental neutron scattering data from Bai et al.
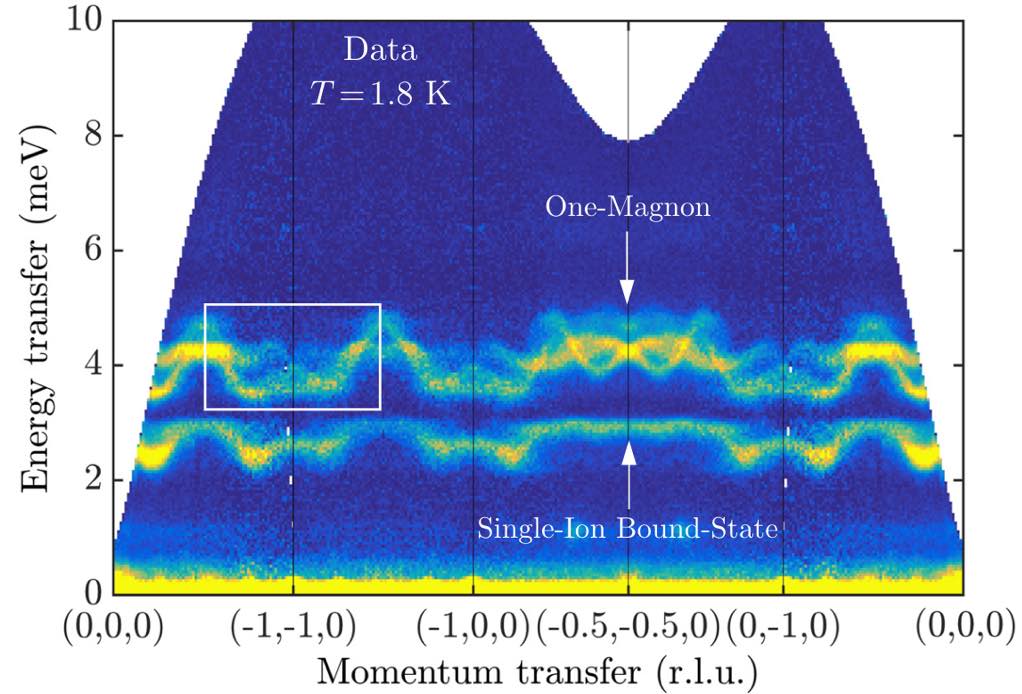
(The publication figure accidentally used a non-standard coordinate system to label the wave vectors.)
To get this agreement, the use of SU(3) coherent states is essential. In other words, we needed a theory of multi-flavored bosons. The lower band has large quadrupolar character, and arises from the strong easy-axis anisotropy of FeI₂. By setting mode = :SUN, the calculation captures this coupled dipole-quadrupole dynamics.
An interesting exercise is to repeat the same study, but using mode = :dipole instead of :SUN. That alternative choice would constrain the coherent state dynamics to the space of dipoles only.
The full dynamical spin structure factor (DSSF) can be retrieved as a $3×3$ matrix with the dssf function, for a given path of $𝐪$-vectors.
disp, is = dssf(swt, path);The first output disp is identical to that obtained from dispersion. The second output is contains a list of $3×3$ matrix of intensities. For example, is[q,n][2,3] yields the $(ŷ,ẑ)$ component of the structure factor intensity for nth mode at the qth wavevector in the path.
The multi-boson linear spin wave theory, applied above, can be understood as the quantization of a certain generalization of the Landau-Lifshitz spin dynamics. Rather than dipoles, this dynamics takes places on the space of SU(N) coherent states.
The full SU(N) coherent state dynamics, with appropriate quantum correction factors, can be useful to model finite temperature scattering data. In particular, it captures certain anharmonic effects due to thermal fluctuations. See our generalized spin dynamics tutorial.
The classical dynamics is also a good starting point to study non-equilibrium phenomena. Empirical noise and damping terms can be used to model coupling to a thermal bath. This yields a Langevin dynamics of SU(N) coherent states. Our dynamical SU(N) quench tutorial illustrates how a temperature quench can give rise to novel liquid phase of CP² skyrmions.
Settings
This document was generated with Documenter.jl version 1.2.1 on Wednesday 3 January 2024. Using Julia version 1.9.4.