Author: Noah Spies
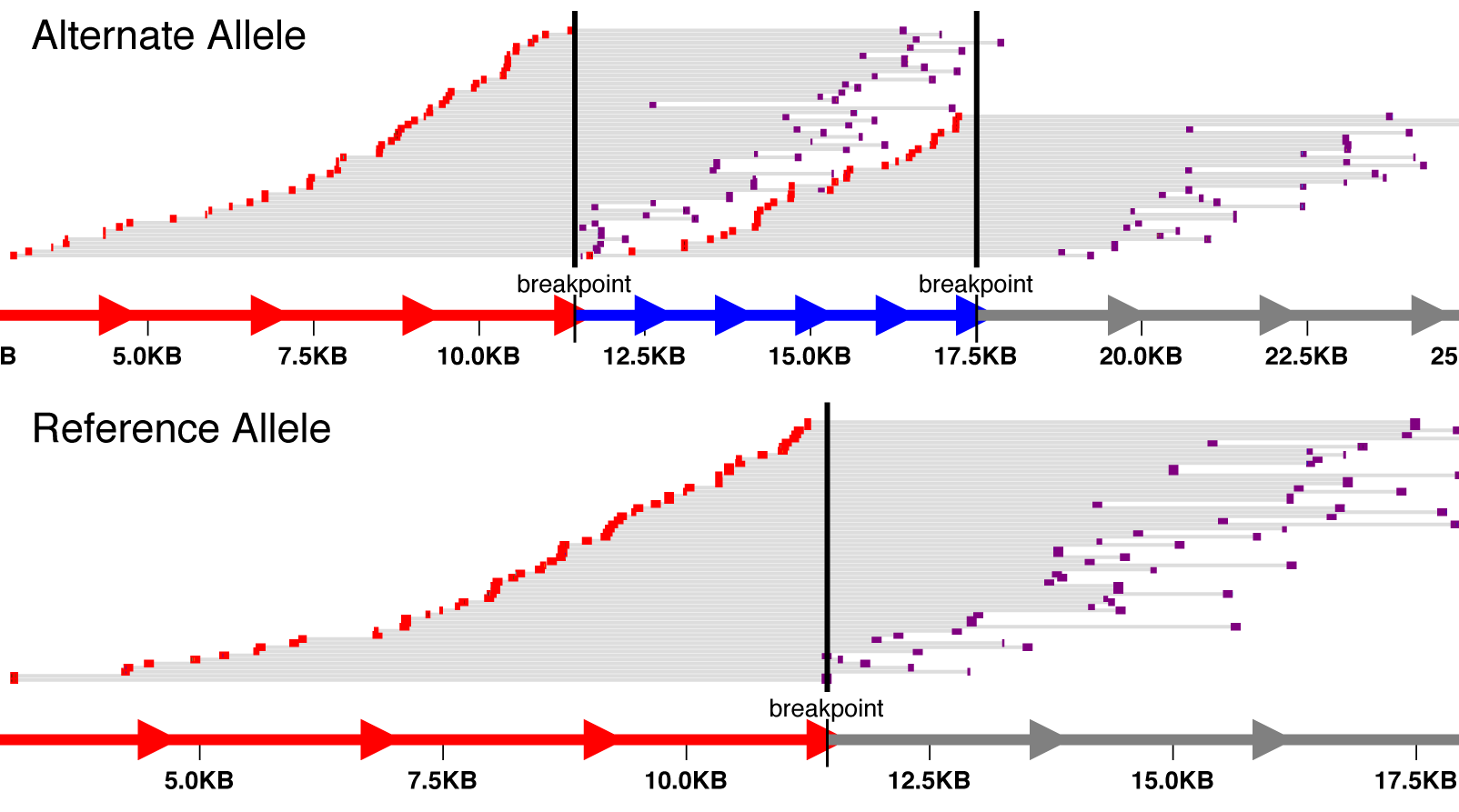
svviz visualizes high-throughput sequencing data relevant to a structural variant. Only reads supporting the variant or the reference allele will be shown. svviz can operate in both an interactive web browser view to closely inspect individual variants, or in batch mode, allowing multiple variants (annotated in a VCF file) to be analyzed simultaneously.
Visit the project site for a tour of the features and example output.
svviz has been tested on Mac OS X and Linux, and should work on Windows, although that has not been tested.
- Ensure that you have a working compiler by following these instructions (OS X only) and that python and the python package pip are installed.
- Install the latest version of svviz from github using the following terminal command:
sudo pip install -U svviz. (The sudo may not be necessary depending on your setup.) - Run the following command, which downloads example data and runs it through svviz:
svviz demo. After several processing steps, a web browser window should open, with separate, interactive views of the reference and alternate alleles.
See the documentation for more detailed help, or run svviz -h to get help on command line arguments.
Please submit bug reports or feature requests on the github issue tracker.
svviz has been published in Bioinformatics. If you found svviz useful for your research, please cite svviz as follows:
Spies N, Zook JM, Salit M, Sidow A. 2015. svviz: a read viewer for validating structural variants. Bioinformatics doi:bioinformatics/btv478.