This is a minor release aimed towards a nextclade dataset upgrade from 2022-10-27 to 2023-01-09 which adds nomenclature for newly designated recombinants XBH - XBP. This release also adds initial support for the detection of "recursive recombination" including XBL and XBN which are recombinants of XBB.
- Issue #24: Create documentation on Read The Docs
- Issue #210: Handle numeric strain names.
- Issue #185: Simplify creation of the pango-lineage nomenclature phylogeny to use the lineage_notes.txt file and the pango_aliasor library.
- Issue #195: Add bypass to intermission allele ratio for edge cases.
- Issue #204: Add special handling for XBB sequenced with ARTIC v4.1 and dropout regions.
- Issue #205: Add new column
parents_conflictto indicate whether the reported lineages from covSPECTRUM conflict with the reported parental clades from `sc2rf. - Issue #213: Add
XBKto auto-pass lineages. - Issue #222: Add new parameter
--gisaid-access-keytosc2rfandsc2rf_recombinants. - Issue #229: Fix bug where auto-pass lineages are missing when exclude_negatives is set to true.
- Issue #231: Fix bug where 'null' lineages in covSPECTRUM caused error in
sc2rfpostprocess. - The order of the
postprocessing.pywas rearranged to have more comprehensive details for auto-pass lineages. - Add
XANto auto-pass lineages.
- Issue #209: Restrict the palette for
rbd_levelto the range of0:12. - Issue #218: Fix bug concerning data fragmentation with large numbers of sequences.
- Issue #221: Remove parameter
--singletonsin favor of--min-cluster-sizeto control cluster size in plots. - Issue #224: Fix bug where plot crashed with extremely large datasets.
- Combine
plotandplot_historicalinto one snakemake rule. Also at custom patternplot_NX(ex.plot_N10) to adjust min cluster size.
- Combine
reportandreport_historicalinto one snakemake rule.
- Issue #225: Fix bug where false negatives passed validation because the status column wasn't checked.
- Issue #217:
XBB.1.5 - Issue #196:
XBF - Issue #206:
XBG - Issue #196:
XBH - Issue #199:
XBJ - Issue #213:
XBK - Issue #219:
XBL - Issue #215:
XBM - Issue #197:
XBN
- Issue #203:
proposed1305 - Issue #208:
proposed1340 - Issue #212:
proposed1425 - Issue #214:
proposed1440 - Issue #216:
proposed1444 - Issue #220:
proposed1576
2964b4a1docs: update notes to include 1576 proposed issuefdc874abdocs: add test summary package for v0.7.03f3d4438docs: update docs v0.7.078696b36script: add bug fix to sc2rf postprocess for #231403777a0script: lint plotting script2a09c783script: fix sc2rf postprocess bug in duplicate removald44d5f90data: add XBP to controls-gisaid4293439cprofile: add controls-gisaid to virusseq builds91d6fb89defaults: update nextclade dataset to 2023-02-01630b2cd5resources: update49e6f598profile: add virusseq profile7e586d1dscript: add extra logic for auto-passing lineages0ebe5e9cscript: fix bug in report where it didn't check that plots existed25b2f243docs: update developers guide914d933fdefaults: add XBN to controls-gisaid and validation8eaf08a9data: restore controls-gisaid strain listfa123009script: defragment plot for 2185f24f695dataset: update controls-gisaid strain listefc5aab7defaults: update validation to fix XBH dropout5a76f81csc2rf: update sc2rf to use gisaid access token84a01cfcscript: fix validate bug for #225cc031e37env: version control all docs dependenciese1c18b91env: add kaleido for static image exportf63a254adocs: update configuration and faq41c533fddocs: update links in developers guide71d48037defaults: update validation values35a5d1eescript: change slurm job name to basename of conda env04e425f8docs: change links to html refsccda5121docs: add pipeline definiton example18e7ae78docs: remove README content and link to read the docs5f7ec1a1docs: rename sphinx pagesd9cc87ddworkflow: create new pattern plot_NX to customize min cluster size1c3cc7a4docs: success with read the docs, fix description include5921240benv: add sphinx rtd themecd789917docs: try read the docs with requirements6d63bcbfdocs: remove sphinx conda too slow47fe5a54env: switch git from anaconda to conda-forge channel7e8c4c6cdocs: downgrade sphinx python to 3.828e7be0edocs: attempt conda 3 with sphinx readthedocs985159b6docs: attempt conda 2 with sphinx readthedocs7ab1c5d9docs: attempt conda with sphinx readthedocs6d7079a8docs: add sphinx for readthedocs #241809dad5workflow: update validation for new controls-gisaid strains71c0fb34script: catch tight layout warnings for #224690bbd7cresources: update issues348264b4resources: add proposed1444 and proposed1576 to breakpoints3f657060resources: add XBL and XBM to breakpointsa8528cc0docs: add development section to READMEb0446f66docs: add development section to README03a65ebfci: extend miniforge-variant to all jobs for #223f186a1c6ci: use miniforge-variant to fix #223cb1bda73script: add intermission ratio bypass for #195c0429ec3parameter: update nextclade data2377fce6script: improve postprocess duplicate reconciliation for lineage matches6b891c92resources: add proposed1440 to breakpoints #2147171dcfdresources: add proposed1425 to breakpoints #212d5a2bf1cresources: add proposed1393 to breakpoints #21118961b51script: improve postprocess duplicate reconciliation with multiple strain matches1d7263f9resources: add proposed1340 to breakpoints #2089b06cff6resources: update validation for controls762fc77dscript: fix postprocess bug in auto-pass parsingea7cdcafparam: add XBK to resources and vocccc08dbaresources: upgrade sc2rf, upgrade Nextclade #176, update XBG breakpoints #206, parent conflict #205, rbd palette #209c0c9209bscript: fix cluster str recode in plot_breakpoints044040c2param: add XAN to auto-passd7e728d0script: handle numeric strain names for #210c014cfb7script: hardcode rbd palette range from 0 to 12 for #20991f4bb05script: fix missing X parent in lineage_tree for #185f341dd59ci: switch flake8 repo from gitlab to githubdea9eeb3resources: update XBB validation lineagebe32e69eworkflow: fix typo in nextclade dataset_dir575255b8workflow: move lineage tree to resources as not dependent on nextclade for #185447b0b8benv: add pango-aliasor for #185823428a9param: add XBB_ARTIC alt breakpoints for #204
This is a minor bugfix release aimed towards resolving network connectivity errors and catching false positives.
- Issue #195: Consider alleles outside of parental regions as intermissions (conflicts) to catch false positives.
- Issue #201: Make LAPIS query of covSPECTRUM optional, to help with users with network connectivity issues. This can be set with the flag
lapis: falsein builds under the rulesc2rf_recombinants. - Issue #202: Document connection errors related to LAPIS and provide options for solutions.
83ee0139docs: update changelog for v0.6.100fe2fc8docs: update notes for v0.6.1fa03ea96workflow: fix bug where rbd_levels log was incorrectly nameda281b75cworkflow: make lapis optional param for #201 #20275684b55docs: update docs1085ce0escript: postprocess count alleles outside regions as intermissions for #195c11770c1param: add XAV to auto-pass for #104 #195
This is a major release that includes the following changes:
-
Detection of all recombinants in Nextclade dataset 2022-10-27:
XAtoXBE. -
Implementation of recombinant sublineages (ex.
XBB.1). -
Implementation of immune-related statistics (
rbd_level,immune_escape,ace2_binding) fromnextclade, theNextstrainteam, and Jesse Bloom's group:- https://github.com/nextstrain/ncov/blob/master/defaults/rbd_levels.yaml
- https://jbloomlab.github.io/SARS-CoV-2-RBD_DMS_Omicron/epistatic-shifts/
- https://jbloomlab.github.io/SARS2_RBD_Ab_escape_maps/escape-calc/
- https://doi.org/10.1093/ve/veac021
- https://doi.org/10.1101/2022.09.15.507787
- https://doi.org/10.1101/2022.09.20.508745
- Issue #168: NULL collection dates and NULL country is implemented.
controlswas updated to in include 1 strain fromXBBfor a total of 22 positive controls. The 28 negative controls were unchanged fromv0.5.1.controls-gisaidstrain list was updated to includeXAthrough toXBEfor a total of 528 positive controls. This includes sublineages such asXBB.1andXBB.1.2which synchronizes with Nextclade Dataset 2022-10-19. The 187 negatives controls were unchanged fromv0.5.1.
- Issue #176: Upgrade Nextclade dataset to tag
2022-10-27and upgrade Nextclade tov2.8.0. - Issue #193: Use the nextclade dataset
sars-cov-2-21Lto calculateimmune_escapeandace2_binding.
- Issue #193: Create new rule
rbd_levelsto calculate the number of key receptor binding domain (RBD) mutations.
- Issue #185: Use nextclade dataset Auspice tree for lineage hierarchy. Previously, the phylogeny of lineages was constructed from the cov-lineages website YAML. Instead, we now use the tree provided with nextclade datasets, to better synchronize the lineage model with the output.
Rather than creating the output tree in resources/lineages.nwk, the lineage tree will output to data/sars-cov-2_<DATE>/tree.nwk. This is because different builts might use different nextclade datasets, and so are dataset specific output.
- Issue #179: Fix bug where
sc2rf/recombinants.ansi.txtis truncated. - Issue #180: Fix recombinant sublineages (ex. XAY.1) missing their derived mutations in the
cov-spectrum_query. Previously, thecov-spectrum_querymutations were only based on the parental alleles (before recombination). This led to sublinaeges (ex.XAY.1,XAY.2) all having the exact same query. Now, thecov-spectrum_querywill include all substitutions shared between all sequences in thecluster_id. - Issue #187: Document bug that occurs if duplicate sequences are present, and the initial validation was skipped by not running
scripts/create_profile.sh. - Issue #191 and Issue #192: Reduce false positives by ensuring that each mode of sc2rf has at least one additional parental population that serves as the alternative hypothesis.
- Issue #195: Implement a filter on the ratio of intermissions to alleles. Sequences will be marked as false positives if the number of intermissions (i.e. alleles that conflict with the identified parental region) is greater than or equal to the number of alleles contributed by the minor parent. This ratio indicates that there is more evidence that conflicts with recombination than there is allele evidence that supports a recombinant origin.
- Issue #183: Recombinant sublineages. When nextclade calls a lineage (ex.
XAY.1) which is a sublineage of a sc2rf lineage (XAY), we prioritize the nextclade assignment. - Issue #193: Add immune-related statistics:
rbd_levels,rbd_substitutions,immune_escape, andace2_binding.
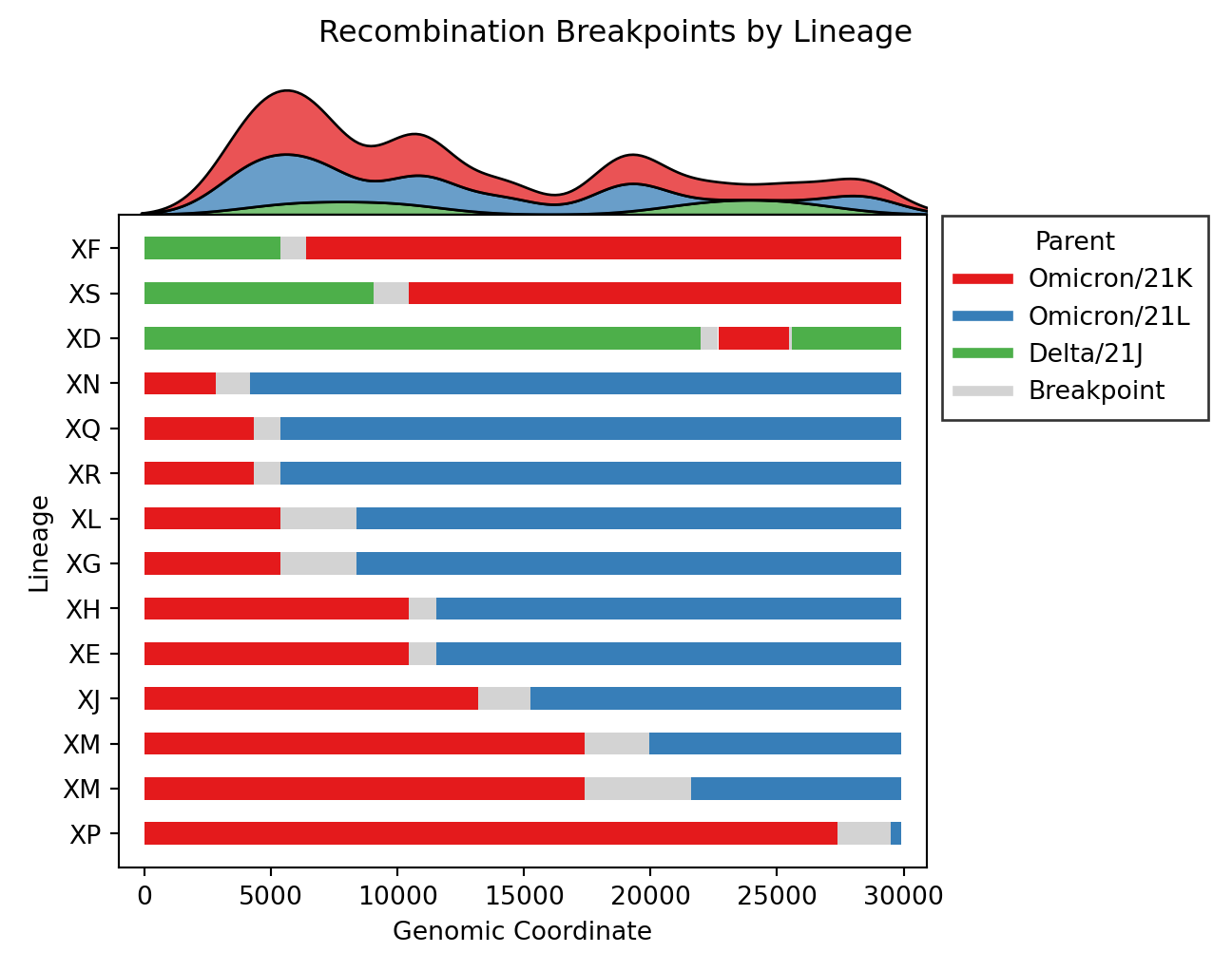
- Issue #57: Include substitutions within breakpoint intervals for breakpoint plots. This is a product of Issue #180 which provides access to all substitutions.
- Issue #112: Fix bug where breakpoints plot image was out of bounds.
- Issue #188: Remove the breakpoints distribution axis (ex.
breakpoints_clade.png) in favor of putting the legend at the top. This significant reduces plotting issues (ex. Issue #112). - Issue #193: Create new plot
rbd_level.
- Issue #85:
XAY, updated controls - Issue #178:
XAY.1 - Issue #172:
XBB.1 - Issue #175:
XBB.1.1 - Issue #184:
XBB.1.2 - Issue #173:
XBB.2 - Issue #174:
XBB.3 - Issue #181:
XBC.1 - Issue #182:
XBC.2 - Issue #171:
XBD - Issue #177:
XBE
- Issue #198:
proposed1229 - Issue #199:
proposed1268 - Issue #197:
proposed1296
2506e907docs: update changelog and add v0.6.0 testing summary package0cc421e0docs: update all contributorscd9b6cbbresources: update issues0fa2e3c1docs: update readme375c3a76resources: add proposed lineages for #197 #198 #199dad989e7param: remove BQ.1 from sc2rf mode VOC as its too close to BA.5.3d7cb005fdocs: update issue template lineage-validation1beac97eresources: add XBF to curated breakpoints for #196fae7bfdbscript: sc2rf implement intermission allele ratio for #19589a41265script: additional manual curation of lineage_treeebd3ce1fresources: update validation strains for controls-gisaidd8bff572script: add RBD Level slide to reportc1879c1dscript: catch errors in rbd_level plotting with no recombinants63545a08script: fix bug in linelist with cluster_privatesc24a7179resources: update issuesd32d557fdocs: update development notes7f825a41script: manual fix for CK in lineage_treefdd6f66dworkflow: implement rbd levels for #1930058dd6eparam: upgrade nextclade dataset to 2022-10-27 and reduce breakpoints of XA modefb062c32env: upgrade nextclade to v2.8.0800c1e9cparam: experiment with XAJ mode for 1916958337fenv: version control pip to v22.3 on conda-forge2bf5141benv: change anaconda channel to conda-forge for #1709b484d3bscript: allow NULL dates in metadata for #168 remove breakpoint dist axis for #188 fix breakpoints plot out of bounds for #11290b153e7docs: update dev notes5057814aresources: remove deprecated lineages and geo_resolutions28c1b8bdresources: update issues and breakpoints379112b4profile: update validation valuesf8f4273cprofile: update controls-gisaid to XBEe462ceadprofile: add XBB.1 to controlsc2aca577profile: increase max jobs in controls-gisaid-hpceabbed42param: add more BA.5 sublineages to sc2rf lineages75e674e0workflow: implement sublineages for #1835caaaa4fworkflow: use nextclade dataset for phylogeny for #185732f5eearesources: add XBC.1 to breakpoints for #1812c1f09a5resources: add new lineage XAY.1 to curated breakpoints for #178b44e68e1script: use all substitutions for cov-spectrum for #180 and #579d222e1fscript: ignore issue 848 when downloading, is manually curated98c8a912script: bugfix where sc2rf ansi was truncated for #1791e29d7eaenv: upgrade nextclade and nextclade dataset for #176409183f5resources: update breakpoints for proposed1139 #165
- Issue #169: AttributeError: 'str' object has no attribute 'name'
- Issue #167: Alias key out of date, change source
- Issue #166:
proposed1138 - Issue #165:
proposed1139
799904ebdocs: update CHANGELOG for v0.5.11f9cd623docs: update docs for v0.5.143fc4d71env: update tabulate channel for #1693b9c3796env: version control tabulate for #1695f57bca2script: update alias url for #16731647371docs: update readme hpc section
Please check out the
v0.5.0Testing Summary Package for a comprehensive report.
This is a minor release that includes the following changes:
- Detection of all recombinants in Nextclade dataset 2022-09-27:
XAtoXBC. - Create any number of custom
sc2rfmodes with CLI arguments.
- Issue #96: Create newick phylogeny of pango lineage parent child relationships, to get accurate sublineages including aliases.
- Issue #118: Fix missing pango-designation issues for XAY and XBA.
- Issue #25: Reduce positive controls to one sequence per clade. Add new positive controls
XAL,XAP,XAS,XAU, andXAZ. - Issue #92: Reduce negative controls to one sequence per clade. Add negative control for
22D (Omicron) / BA.2.75. - Issue #155: Add new profile and dataset
controls-gisaid. Only a list of strains is provided, as GISAID policy prohibits public sharing of sequences and metadata.
- Issue #77: Report slurm command for
--hpcprofiles inscripts/create_profiles.sh. - Issue #153: Fix bug where build parameters
metadataandsequenceswere not implemented.
-
Issue #78: Add new parameter
max_breakpoint_lentosc2rf_recombinantsto mark samples with two much uncertainty in the breakpoint interval as false positives. -
Issue #79: Add new parameter
min_consec_alleletosc2rf_recombinantsto ignore recombinant regions with less than this number of consecutive alleles (both diagnostic SNPs and diganostic reference alleles). -
Issue #80: Migrate sc2rf froma submodule to a subdirectory (including LICENSE!). This is to simplify the updating process and avoid errors where submodules became out of sync with the main pipeline.
-
Issue #83: Improve error handling in
sc2rf_recombinantswhen the input stats files are empty. -
Issue #89: Reduce the default value of the parameter
min_leninsc2rf_recombinantsfrom1000to500.This is to handleXAPandXAJ. -
Issue #90: Auto-pass select nextclade lineages through
sc2rf:XN,XP,XAR,XAS, andXAZ. This requires differentiating the nextclade inputs as separate parameters--nextcladeand--nextclade-no-recom.XN,XP, andXARhave extremely small recombinant regions at the terminal ends of the genome. Depending on sequencing coverage,sc2rfmay not reliably detect these lineages.The newly designated
XASandXAZpose a challenge for recombinant detection using diagnostic alleles. The first region ofXAScould be eitherBA.5orBA.4based on subsitutions, but is mostly likelyBA.5based on deletions. Since the region contains no diagnostic alleles to discriminateBA.5vs.BA.4, breakpoints cannot be detected bysc2rf.Similarly for
XAZ, theBA.2segments do not contain anyBA.2diagnostic alleles, but instead are all reversion fromBA.5alleles. TheBA.2parent was discovered by deep, manual investigation in the corresponding pango-designation issue. Since theBA.2regions contain no diagnostic forBA.2, breakpoints cannot be detected bysc2rf. -
Issue #95: Generalize
sc2rf_recombinantsto take any number of ansi and csv input files. This allows greater flexibility in command-line arguments tosc2rfand are not locked into the hardcodedprimaryandsecondaryparameter sets. -
Issue #96: Include sub-lineage proportions in the
parents_lineage_confidence. This reduces underestimating the confidence of a parental lineage. -
Issue #150: Fix bug where
sc2rfwould write empty output csvfiles if no recombinants were found. -
Issue #151: Fix bug where samples that failed to align were missing from the linelists.
-
Issue #158: Reduce
sc2rfparam--max-intermission-lengthfrom3to2to be consistent with Issue #79. -
Issue #161: Implement selection method to pick best results from various
sc2rfmodes. -
Issue #162: Upgrade
sc2rf/virus_properties.json. -
Issue #163: Use LAPIS
nextcladePangoLineageinstead ofpangoLineage. Also disable default filtermax_breakpoint_lenforXAN. -
Issue #164: Fix bug where false positives would appear in the filter
sc2rfansi output (recombinants.ansi.txt). -
The optional
lapisparameter forsc2rf_recombinantshas been removed. Querying LAPIS for parental lineages is no longer experimental and is now an essential component (cannot be disabled). -
The mandatory
mutation_thresholdparameter forsc2rfhas been removed. Instead,--mutation-thresholdcan be set independently in each of thescrfmodes.
- Issue #157: Create new parameters
min_lineage_sizeandmin_private_mutsto control lineage splitting intoX*-like.
- Issue #17: Create script to plot lineage assignment changes between versions using a Sankey diagram.
- Issue #82: Change epiweek start from Monday to Sunday.
- Issue #111: Fix breakpoint distribution axis that was empty for clade.
- Issue #152: Fix file saving bug when largest lineage has
/characters.
- Issue #88: Add pipeline and nextclade versions to powerpoint slides as footer. This required adding
--summaryas param toreport.
- Issue #56: Change rule
validatefrom simply counting the number of positives to validating the fieldslineage,breakpoints,parents_clade. This involves adding a new default parameterexpectedfor rulevalidateindefaults/parameters.yaml.
- Issue #149:
XA - Issue #148:
XB - Issue #147:
XC - Issue #146:
XD - Issue #145:
XE - Issue #144:
XF - Issue #143:
XG - Issue #141:
XH - Issue #142:
XJ - Issue #140:
XK - Issue #139:
XL - Issue #138:
XM - Issue #137:
XN - Issue #136:
XP - Issue #135:
XQ - Issue #134:
XR - Issue #133:
XS - Issue #132:
XT - Issue #131:
XU - Issue #130:
XV - Issue #129:
XW - Issue #128:
XY - Issue #127:
XZ - Issue #126:
XAA - Issue #125:
XAB - Issue #124:
XAC - Issue #123:
XAD - Issue #122:
XAE - Issue #120:
XAF - Issue #121:
XAG - Issue #119:
XAH - Issue #117:
XAJ - Issue #116:
XAK - Issue #115:
XAL - Issue #110:
XAM - Issue #109:
XAN - Issue #108:
XAP - Issue #107:
XAQ - Issue #87:
XAS - Issue #105:
XAT - Issue #103:
XAU - Issue #104:
XAV - Issue #105:
XAW - Issue #85:
XAY - Issue #87:
XAZ - Issue #94:
XBA - Issue #114:
XBB - Issue #160:
XBC
- Issue #99:
proposed808
pull/113docs: add issues template for lineage validation
b48ad6d7docs: fix CHANGELOG pr04b17918docs: update readme and changelog72dd5a8fdocs: add testing summary package for v0.4.2 to v0.5.0558f7d79resources: fix breakpoints for XAE #12291e5843bscript: bugfix sc2rf ansi output for #1649bc13639docs: update issues and validation table orderb63520e5script: implement lineage check in dups for #117 #161901898dasc2rf updates for #158 #161 #162 #16396fa6af1dataset: update controls-gisaid strain list and validation84466a10workflow: new param dup_method for #1619ca0c71escript: implement duplicate reconciliation for #161112ea684param: upgrade nextclade dataset for #159859b92c8script: add more detail to validate table for failing samples5e285912script: add param --min-link-size to compare_positivesbd01a5e4workflow: added failed validate output to rule log8e5b90fbworkflow: don't use metadata for sc2rf_recombinants when exclude_negatives is truecdf45407param: add new params min-lineage-size and min-private-muts for #157bc04fddfworkflow: update validation strains for #1556aa95221param: fix typo of missing --mutation-threshold25df848cparam: remove param mutation_threshold as universal param for sc2rf46d2ee95dataset: remove false positive LC0797902 from negative controlsb106b9d1profile: change default hpc jobs from 2 to 10d6d02721workflow: update validation tabled308d54bscript: fix node ordering in compare_positivesea65c6ffci: remove GISAID workflow for #154122e579cci: test storing csv files as secrets for #154d281add4ci: experiment with secrets with test data for #1541d5406f5script: generalize compare_positives to use other lineage columnsffd4f159scripts: fix bug where metadata and sequences param were not implemented for #1537974860aresources: special handling of proposed808 issues and breakpoints for #99d0e3f41ascript: fix file saving bug in report for #152f7d3157bscript: fix file saving bug in plot for #152f9931bb3script: fix missing samples in sc2rf output for #15118b94df5script: force sc2rf to always output csvfile headers for #150e1f14dferesources: update breakpoints for proposed808 #9935aaa922resources: update breakpoints for XA - XAZ05cba895resources: update breakpoints for XV #1301b0a02bfresources: add gauntlet samples (all XA*) to validation29c7798dparam: add XAR to sc2rf auto-pass for 1060e6b413edocs: change next ver from v0.4.3 to v0.5.0f8197e80workflow: fix bug in rule validate where path was hard-codedabb4dec6resources: update breakpoints for XAA #126d012b936resources: update breakpoints for XAG and XAH for #120 and #121d389e048param: add new XAJ mode for sc2rf for #117be853ac1scripts: update rule validate for #56a02b4debdocs: add issues template for lineage validation #1131b3cb780script: fix bug of missing issues for #11882d9ce32docs: update validation release notes1b705a6aresources: update XAU breakpoints for #10318021fe3docs: add XAQ issue #107 to release notes7ca8f06fdocs: add XAQ issue #107 to release notese9d8f905docs: add issue #111 to release notesa638835ascript: fix bug in plot_breakpoints when axis empty for #111ba7ec30cresources: update breakpoints for XAP #108d3952e44docs: fix typo in relesae notesd56956d6docs: add issues #86 and #87 to release notes00a706a8script: remove redundant --clades arg in sc2rf bash script378eea62param: add new sc2rf modes XB and proposed808 for #98 and #99bb802647docs: add issue #17 to release notesb6688c86env: add plotly to conda env and control all versionsadc6777escript: improve directory creation in compare positives15a1fc04script: add breakpoint axis label for #97f256a75fdocs: add notes for v0.4.3c3785fa0env: upgrade csvtk to v0.24.07936e0efparam: fix typo in mode omicron_omicrond014d0a1param: revert XAS mode to default for #86ea83d78escript: fix bug in postprocess where max_breakpoint_len was not checkedab030e21param: add new XAS mode to default sc2rf runs for #866ccd0aa8workflow: first draft of pango lineage tree for #969d2382cbworkflow: add param fix for postprocess inputs93007726script: fix cli --clades arg parsing for scr2rf.shbed29fdbscript: add new csv col alleles to sc2rfbd681d5eworkflow: generalize sc2rf_recombinants inputs for #950315ffd6docs: update development docs70111447resources: update breakpoints and issues076e14bbdataset: reduce controls to one sequence per clade for #25,9277d2210dworkflow: update rules for #46, #88, #89, #90d84205a0script: add new param auto_pass for #9099b895a4params: update params for #46, #89, #903e3d1022script: add pipeline version to report for #882e6ac558script: remove sc2rf_ver col from summary for #8018c35940env: upgrade nextclade to v2.5.0 for #91a67e1159workflow: autopass XAS through sc2rf for #862e16dac5resources: update breakpoints and mutationsa7026450workflow: upgrade nextclade dataset to 2022-08-23 for #8153fb4a8aworkflow: re-add sc2rf as subdirectory for #804b8b3fabworkflow: remove sc2rf submodule againb650e562workflow: add sc2rf as subdirectory for #80073f3b94workflow: remove sc2rf as submodule5ab5c1b5resources: updated curated breakpoints5635b872resources: update issues78e1f064script: change epiweek to start on Sundary (cdc) for #8237f40480script: add tables to compare positives between versions for #178eef7548script: create new script to compare positives between versionsebf1e222script: compare linelists from different versions for #178401c353workflow: add new param max_breakpoint_len for #788bbcc041script: report slurm command for --hpc profiles for #77c40a6791workflow: restrict config rules to one threadc2b1ea57script: revert unpublished lineages for #76dbe359c8resources: add 882 to breakpoints
This is a minor bug fix and enhancement release with the following changes:
- Issue #70: Fix missing
sc2rfversion fromrecombinant_classifier_dataset - Issue #74: Correctly identify
XN-likeandXP-like. Previously, these were just assignedXN/XPregardless of whether the estimated breakpoints conflicted with the curated ones. - Issue #76: Mark undesignated lineages with no matching sc2rf lineage as
unpublished.
- Issue #71: Only truncate
cluster_idwhile plotting, not in table generation. - Issue #72: For all plots, truncate the legend labels to a set number of characters. The exception to this are parent labels (clade,lineage) because the full label is informative.
- Issue #73, #75: For all plots except breakpoints, lineages will be defined by the column
recombinant_lineage_curated. Previously it was defined by the combination ofrecombinant_lineage_curatedandcluster_id, which made cluttered plots that were too difficult to interpret. - New parameter
--lineage-colwas added toscripts/plot_breakpoints.pyto have more control on whether we want to plot the raw lineage (lineage) or the curated lineage (recombinant_lineage_curated).
8953ef03docs: add CHANGELOG for v0.4.27ec5ccc6docs: add notes for v0.4.21b3b1f1dscript: restore column name to recombinant_classifer_dataset901caf98script: restore recombinant_lineage_curated of -like lineagesd6be9611script: change internal delim of classifier for #70cdb4a78ascript: fix recombinant_classifier missing sc2rf for #70bf7a4e57script: mark undesignated lineages with no matching sc2rf lineage as unpublished for #7646f6d754workflow: update linelists and plotting for #74 and #75c03dd3bescript: don't split largest by cluster id for #73e9802e79script: majority of plots will not split by cluster_id for #73bafb38fbscript: fix cluster ID truncation for issue #71ab712593resources: curate and test breakpoints for proposed895
This is a minor bug fix release with the following changes:
- Issue #63: Remove
usherandprotobuffrom the conda environment. - Issue #68: Remove ncov as a submodule.
- Issue #69: Remove 22C and 22D from
sc2rf/mapping.csvandsc2rf/virus_properties.json, as these interfere with breakpoint detection for XAN.
88650696docs: add CHANGELOG for v0.4.100a2eec3docs: add notes for v0.4.1d74a81d3sc2rf: revert 22C and 22D clade addition7b662940env: remove usher for issue #63adf92399submodule: remove ncov for issue #680790aa04docs: update CHANGELOG for v0.4.0
v0.4.0 has been trained and validated on the latest generation of SARS-CoV-2 Omicron clades (ex. 22A/BA.4 and 22B/BA.5). Recombinant sequences involving BA.4 and BA.5 can now be detected, unlike in v0.3.0 where they were not included in the sc2rf models.
v0.4.0 is also a major update to how sequences are categorized into lineages/clusters. A recombinant lineage is now defined as a group of sequences with a unique combination of:
- Lineage assignment (ex.
XM) - Parental clades (ex.
Omicron/21K,Omicron/21L) - Breakpoints (ex.
17411:21617) - NEW: Parental lineages (ex.
BA.1.1,BA.2.12.1)
Novel recombinants (i.e. undesignated) can be identified by a lineage assignment that does not start with X* (ex. BA.1.1) or with a lineage assignment that contains -like (ex. XM-like). A cluster of sequences may be flagged as -like if one of the following criteria apply:
-
The lineage assignment by Nextclade conflicts with the published breakpoints for a designated lineage (
resources/breakpoints.tsv).- Ex. An
XEassigned sample has breakpoint11538:12879, which conflicts with the publishedXEbreakpoint (ex. 8394:12879). This will be renamedXE-like.
- Ex. An
-
The cluster has 10 or more sequences, which share at least 3 private mutations in common.
- Ex. A large cluster of sequences (N=50) are assigned
XM. However, these 50 samples share 5 private mutationsT2470C,C4586T,C9857T,C12085T,C26577Gwhich do not appear in trueXMsequences. These will be renamedXM-like. Upon further review of the reported matching pango-designation issues (460,757,781,472,798), we find this cluster to be a match toproposed798.
- Ex. A large cluster of sequences (N=50) are assigned
The ability to identify parental lineages and private mutations is largely due to improvements in the newly released nextclade datasets, , which have increased recombinant lineage accuracy. As novel recombinants can now be identified without the use of the custom UShER annotations (ex. proposed771), all UShER rules and output have been removed. This significantly improves runtime, and reduces the need to drop non-recombinant samples for performance. The result is more comparable output between different dataset sizes (4 samples vs. 400,000 samples).
Note! Default parameters have been updated! Please regenerate your profiles/builds with:
scripts/create_profile.sh --data data/custom
- Issue #49: The tutorial lineages were changed from
XM,proposed467,miscBA1BA2Post17k, toXD,XH,XAN. The previous tutorial sequences had genome quality issues. - Issue #51: Add
XANto the controls dataset. This is BA.2/BA.5 recombinant. - Issue #62: Add
XAKto the controls dataset. This is BA.2/BA.1 VUM recombinant monitored by the ECDC.
- Issue #46:
nextcladeis now run twice. Once with the regularsars-cov-2dataset and once with thesars-cov-2-no-recombdataset. Thesars-cov-2-no-recombdataset is used to get the nucleotide substitutions before recombination occurred. These are identified by taking thesubstitutionscolumn, and excluding the substitutions found inprivateNucMutations.unlabeledSubstitutions. The pre-recombination substitutions allow us to identify the parental lineages by querying cov-spectrum. - Issue #48: Make the
exclude_cladescompletely optional. Otherwise an error would be raised if the user didn't specify any. - Issue #50: Upgrade from
v1.11.0tov2.3.0. Also upgrade the default dataset tags to 2022-07-26T12:00:00Z which had significant bug fixes. - Issue #51: Relax the recombinant criteria, by flagging sequences with ANY labelled private mutations as a potential recombinant for further downstream analysis. This was specifically for BA.5 recombinants (ex.
XAN) as no other columns from thenextcladeoutput indicated this could be a recombinant. - Restrict
nextcladeoutput tofasta,tsv(alignment and QC table). This saves on file storage, as the other default output is not used.
- Issue #51:
sc2rfis now run twice. First, to detect recombination between clades (ex.Delta/21J&Omicron/21K). Second, to detect recombination within Omicron (ex.Omicron/BA.2/21L&Omicron/BA.5/22B). It was not possible to define universal parameters forsc2rfthat worked for both distantly related clades, and the closely related Omicron lineages. - Issue #51: Rename parameter
cladestoprimary_cladesand add new parametersecondary_cladesfor detecting BA.5. - Issue #53: Identify the parental lineages by splitting up the observed mutations (from
nextclade) into regions by breakpoint. Then query the list of mutations in https://cov-spectrum.org and report the lineage with the highest prevalence. - Tested out
--enable-deletionsagain, which caused issues forXD. This confirms that using deletions is still ineffective for defining breakpoints. - Add
B.1.631andB.1.634tosc2rf/mapping.tsvand as potential clades in the default parameters. These are parents forXB. - Add
B.1.438.1tosc2rf/mapping.tsvand as a otential clade in the default parameters. This is a parent forproposed808. - Require a recombinant region to have at least one substitution unique to the parent (i.e. diagnostic). This reduces false positives.
- Remove the debugging mode, as it produced overly verbose output. It is more efficient to rerun manually with custom parameters tailored to the kind of debugging required.
- Change parent clade nomenclature from
Omicron/21Kto the more comprehensiveOmicron/BA.1/21K. This makes it clear which lineage is involved, since it's not always obvious how Nextclade clades map to pango lineages.
- Issue #63: All UShER rules and output have been removed. First, because the latest releases of nextclade datasets (tag
2022-07-26T12:00:00Z) have dramatically improved lineage assignment accuracy for recombinants. Second, was to improve runtime and simplicity of the workflow, as UShER adds significantly to runtime.
- Issue #30: Fixed the bug where distinct recombinant lineages would occasionally be grouped into one
cluster_id. This is due to the new definition for recombinant lineages (see General) section, which now includes parental lineages and have sufficient resolving power. - Issue #46: Added new column
parents_subs, which are the substitutions found in the parental lineages before recombination occurred using thesars-cov-2-no-recombnextclade dataset. Also added new columns:parents_lineage,parents_lineage_confidence, based on queryingcov-spectrumfor the substitutions found inparents_subs. - Issue #53: Added new column
cov-spectrum_querywhich includes the substitutions that are shared by ALL sequences of the recombinant lineage. - Added new column
cluster_privateswhich includes the private substitutions shared by ALL sequences of the recombinant lineage. - Renamed
parentscolumn toparents_clade, to differentiate it from the new columnparents_lineage.
- Issue #4, Issue #57: Plot distributions of each parent separately, rather than stacking on one axis. Also plot the substitutions as ticks on the breakpoints figure.
| v0.3.0 | v0.4.0 |
|---|---|
 |
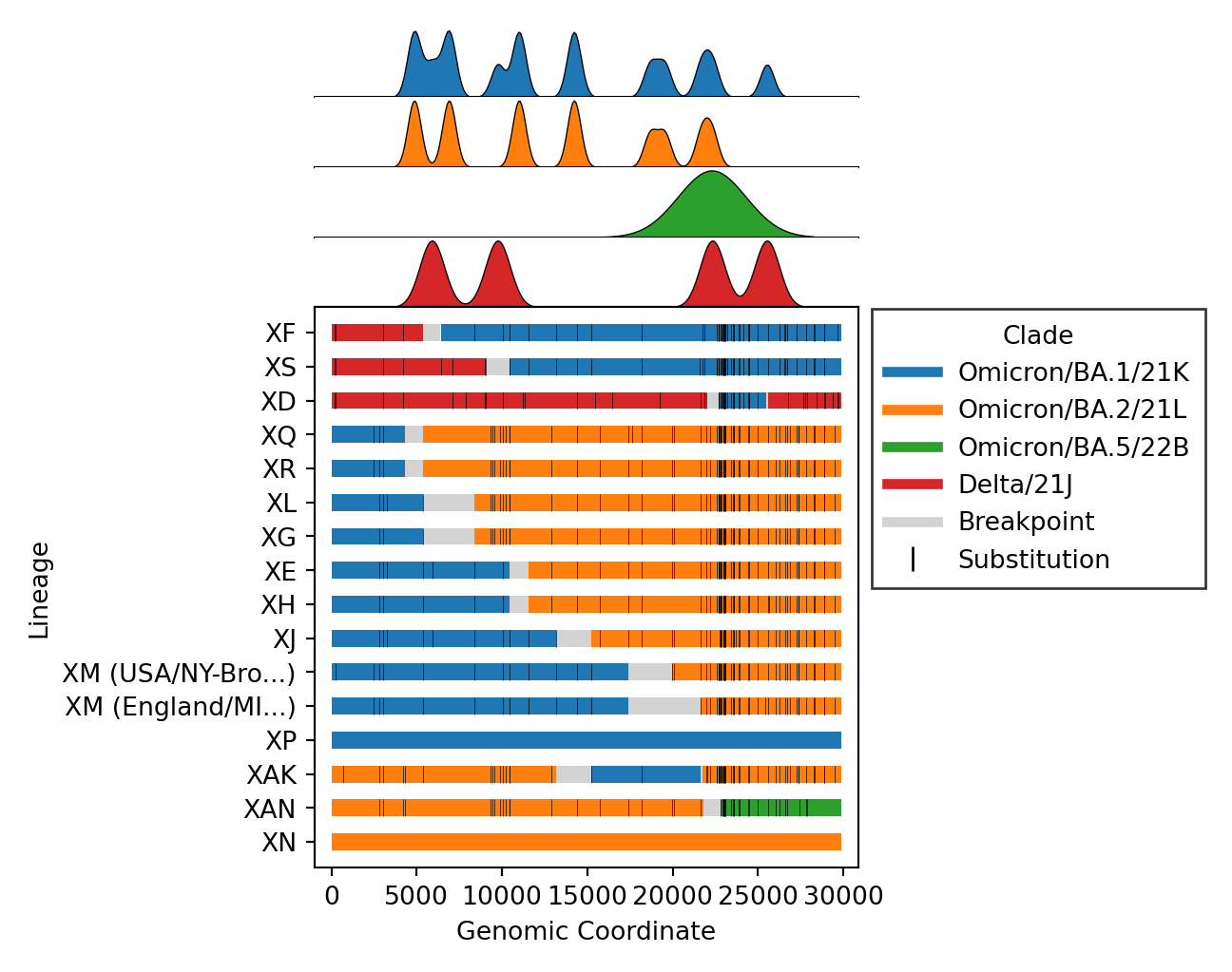 |
- Issue #46: Plot breakpoints separately by clade and lineage. In addition, distinct clusters within the same recombinant lineage are noted by including their cluster ID as a suffix. As an example, please see
XM (USA) and X (England)below. Where the lineage is the same (XM), but the breakpoints differ, as do the parental lineages (BA.2vsBA.2.12.1). These clusters are distinct becauseXM (England)lacks substitutions occurring around position 20000.
| Clade | Lineage |
|---|---|
 |
 |
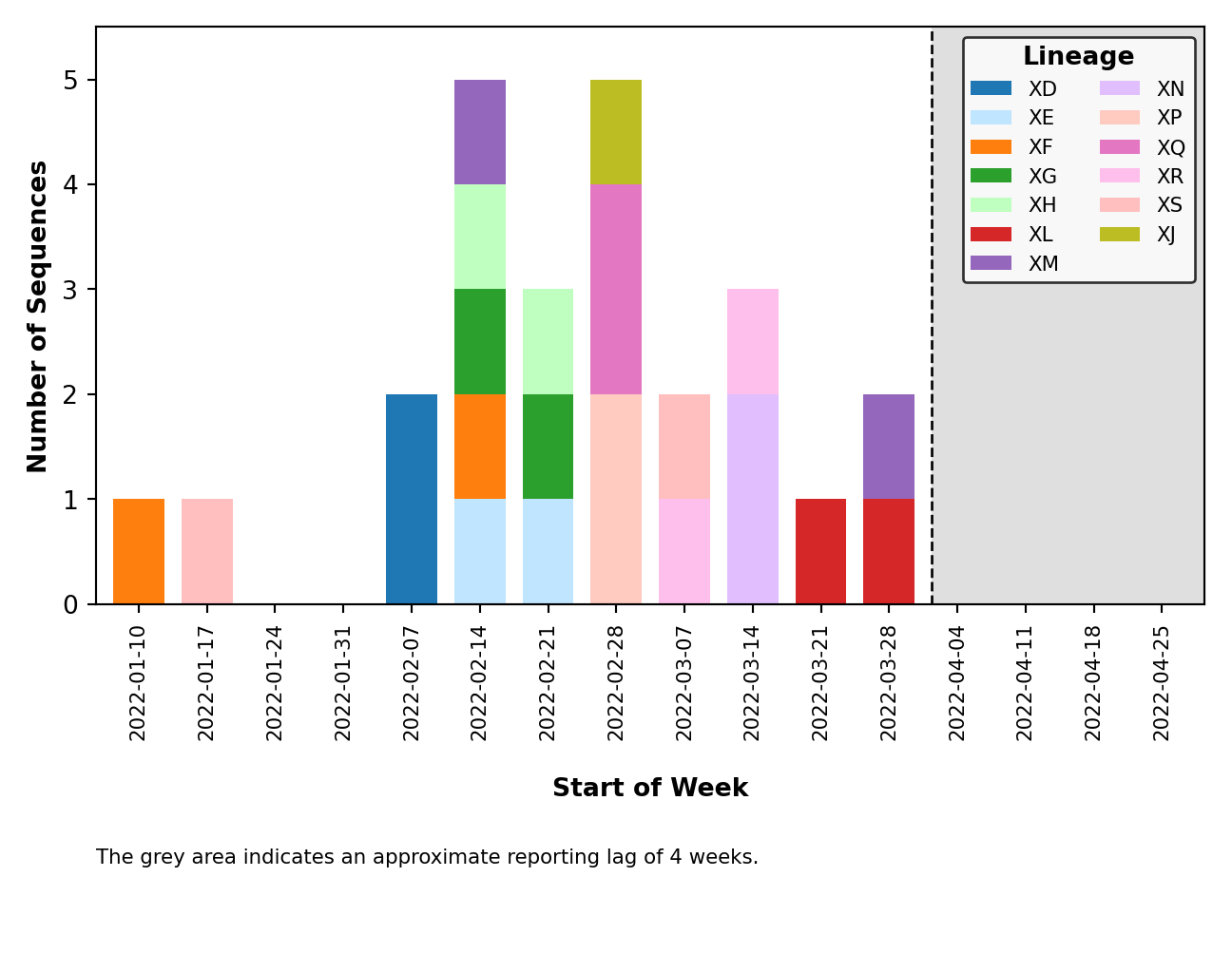
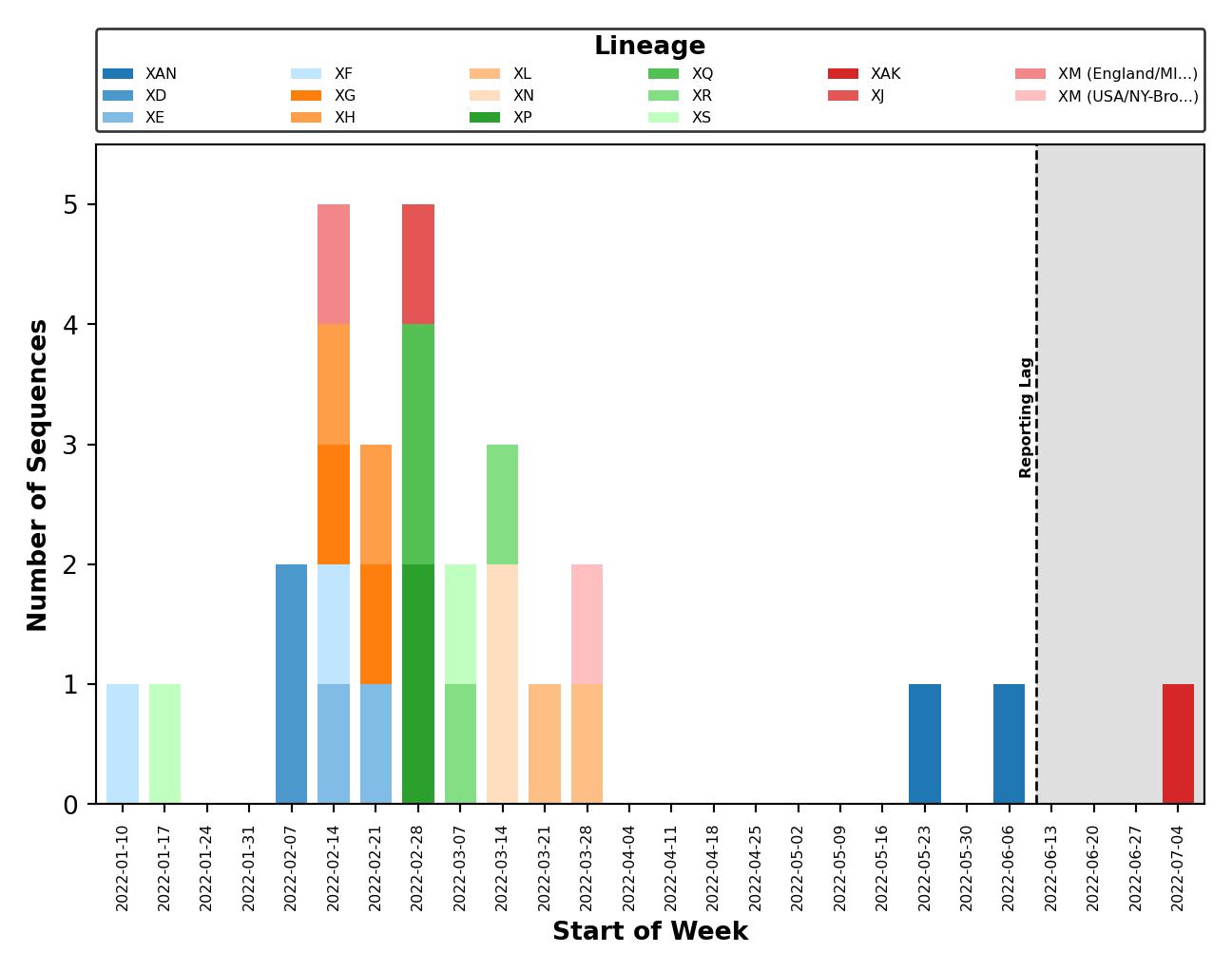
- Issue #58: Fix breakpoint plotting from all lineages to just those observed in the reporting period. Except for the breakpoint plots in
plots_historical. - Issue #59: Improved error handling of breakpoint plotting when a breakpoint could not be identified by
sc2rf. This is possible ifnextcladewas the only program to detect recombination (and thus, we have no breakpoint data fromsc2rf). - Issue #64: Improved error handling for when the lag period (ex. 4 weeks) falls outside the range of collection dates (ex. 2 weeks).
- Issue #65: Improved error handling of distribution plotting when only one sequence is present.
- Issue #67: Plot legends are placed above the figure and are dynamically sized.
| v0.3.0 | v0.4.0 |
|---|---|
 |
 |
- Issue #60: Remove changelog from final slide, as this content did not display correctly
- Issue #61: Fixed bug in the
report.xlsxwhere the number of proposed and unpublished recombinant lineages/sequences was incorrect.
- Issue #58: New rule (
validate) to validate the number of positives in controlled datasets (ex. controls, tutorials) againstdefaults/validation.tsv. If validation fails based on an incorrect number of positives, the pipeline will exit with an error. This is to make it more obvious when results have changed during Continuous Integration (CI)
c027027bdocs: remove dev notes77d9f01bci: remove unit test workflow574f8c15docs: update instructions in README79b61fe2ci: remove unit tests3d53ebd2docs: update notes for v0.4.0b968dc6dscript: add more subs columns to lineages linelist75477ec4script: adjust lag epiweek by 1 when overlaps20409f5csc2rf: mapping change 22C to BA.2.12.1 and add 22D432b6b79docs: add breakpoints lineage output for v0.4.04fbde4b9docs: add breakts output image to compare v0.3.0 and v0.4.0af2f25d3docs: add lineage output image for v0.4.0 to compare5615f113docs: add lineage output image for v0.3.0 to compare57a08096profile: adjust plotting min and max dates for tutorial4226c85bscript: mark nextclade recombinants unverified by sc2rf as false positivesd6700e7adocs: rearrange sections in README7f773ba7docs: update report and slides images and links3599a7f4script: don't use proposed* lineages from sc2rf as consensus lineage76106b50docs: add new image for lineages plot86c2fa2edocs: update README and images488ea6c7script: dynamic legends for #67bec01845script: move plot legend to top for #6742d1281bscript: make sure X*-like lineages have proposed status230f9495resources: update issues3c2f4c84script: detect novel recombinants based on private mutationsd9e279beresources: update breakpoints for XAF and XAG2d070cb2script: improve linelist efficiency for assigning cluster idsb65437f2script: add create_logger function to general functions import489154f6sc2rf: explicitly call variables _primary and _secondary8f464a14script: simplify extra columns for summary script30f90443workflow: formally run sc2rf in primary/secondary mode in parallel6688c828defaults: restore validation counts for XN and XP043b691escript: add special processing for XN and XP939cc967script: only adjust lineage status in linelist if positive28d91363dataset: update tutorial strains for #4909e53a94workflow: remove usher for #6322c63de0workflow: add XAK to controls for Issue #62342fa2a3script: create separate slides for designated, proposed, and unpublished for issue #6184c9b57dscript: create separate plots for designated, proposed, and unpublished for issue #6181c58931script: if lineage is proposed* use as curated lineage rather than cluster_id2db6100bci: fix typo in profile creation job names18102431script: remove changelog from report slides for issue #60b7784b30docs: use new breakpoints path for README66ee7d1dnextclade: upgrade datasets to tag 2022-07-26 for issue #507880327cci: don't trigger pipeline on images changese8c71171docs: update breakpoints imagese13138e8script: rename NA to Unknown parent when plotting breakpointsfc1b6129script: remove unneeded constants in plot77d3614adataset: change controls proposed771 to XANf2d72330script: catch empty plot when using tight_layout for breakpointsb55dbe95workflow: include unpublished in positive status for rule validate557295b5script: plotting catch when breakpoints are NA2306305dprofile: set exclusions for tutorial to defaultc049ccbaresource: update curated breakpoints218bab52env: upgrade nextclade to v2.3.0 for issue #50a9ea0bd4workflow: fix typo in rule validate that hard-coded controlscf613c34workflow: control breakpoint plotting by clusters file0d4b50a4resource: update breakpoints figures6520457ascript: plot subs along with breakpoints for issue #578110faa7script: create a plot for cluster_id mostly for breakpoint plotting6f02f09cscript: for plot import function categorical_palette1f6195dfprofile: by default do not retry jobs2842a4f9workflow: add rule validate for issue #56693b07dfscript: empty df catching in plot breakpoints12567fdeworkflow: classify any sequence with unlabeled private mutations as a potential positive534ac899docs: add more comments to summary script82b6a696script: fix bug in parent palette for plot breakpoints42510719config: remove explicit conda activation in slurm script and profilesab44e5d7docs: update readme contributorse8e2b134dataset: upgrade nextclade dataset086768c7parameters: restrict breakpoints and parents to 10 for sc2rf83af3a73sc2rf: output NA for false positives breakpoints4baee0descript: catch empty dataframe in script plot1dc84bf6profile: adjust plot end date for positive controlsbe431cdfworkflow: restrict nextclade clades again76e31f68resources: add breakpoints by clade07d96a1eworkflow: fix bug in plot_historical2086b254sc2rf: update lineage mapping0d911b20env: reorganize dependenciesc165074bworkflow: separate plot_breakpoints into separate script0497dffdprofile: controls-negative include false positives8bb41c45script: add separate report slides for clade/lineage parent breakpoints411bc235workflow: remove subtree params and add secondary cladesf80386aasc2rf: catch empty secondary csv6d1b03e0sc2rf: add optional secondary csv for #51a629eb06workflow: detect recombination with BA.5 for #51c44b468aenv: remove plotly and kaleido from enva485215ddataset: add proposed771 to controls for #51eba827a9script: define lineages by parental lineages for #46d6319badworkflow: relax nextclade exclusion filter for #48fb7a0a4fenv: upgrade nextclade to v2.2.0 for #5031ec45beresources: update breakpoints parents nomenclature7f00564d(unverified) sc2rf,postprocess: add unique_subs to output3e3e46c2workflow: implement cov-spectrum query to identify parent lineages041de538workflow: customize nextclade to run with our without recombinant dataset7acc598fscript: remove edges from stacked bar plots for issue #43ab589bb7docs: add mark horsman for ideas and design151e481dbug: fix svg font export for issues #42b34c1452bug: fix ouput typo in issues_download601a0c7aresources: also output svg for issues breakpointsa2a8b00dworkflow: add plotting issues breakpoints to rule issues_download5a5a2f41resources: update breakpoints plot2113f6f9script: plot breakpoints of curate lineages in resources5f4b901ddocs: add new breakpoints image to readmebfdf2191workflow: cleanup Thumbs869f3b4escript: add breakpoints as a plot and report slidee988f251bug: fix missing rule_name for _historical8bc6aef4env: add plotly and kaleido to env066e0c00resources: add 781 789 798 to curated breakpoints4ff587c5resources: update issues and curated breakpointscc146425docs: add instructions for updating conda env8592a156bug: fix usher_collapse metadata output to allow for hCoV-19 prefix9ea4f1b3docs: add --recurse-submodules instruction to updating
-
Default parameters have been updated! Please regenerate your profiles/builds with:
bash scripts/create_profile.sh --data data/custom
-
Rule outputs are now in sub-directories for a cleaner
resultsdirectory. -
The in-text report (
report.pptx) statistics are no longer cumulative counts of all sequences. Instead they, will match the reporting period in the accompanying plots.
- Improve subtree collapse effiency (#35).
- Improve subtree aesthetics and filters (#20).
- Fix issues rendering as float (#29).
- Explicitly control the dimensions of plots for powerpoint embedding.
- Remove hard-coded
extra_cols(#26). - Fix mismatch in lineages plot and description (#21).
- Downstream steps no longer fail if there are no recombinant sequences (#7).
- Add new rule
usher_columnsto augment the base usher metadata. - Add new script
parents.py, plots, and report slide to summarize recombinant sequences by parent. - Make rules
plotandreportmore dynamic with regards to plots creation. - Exclude the reference genome from alignment until
faToVcf. - Include the log path and expected outputs in the message for each rule.
- Use sub-functions to better control optional parameters.
- Make sure all rules write to a log if possible (#34).
- Convert all rule inputs to snakemake rule variables.
- Create and document a
create_profile.shscript. - Implement the
--low-memorymode parameter within the scriptusher_metadata.sh.
-
Create new controls datasets:
controls-negativescontrols-positivescontrols
-
Add versions to
genbank_accessionsforcontrols.
- Upgrade UShER to v0.5.4 (possibly this was done in a prior ver).
- Remove
taxoniumandchronumentalfrom the conda env.
-
Add parameters to control whether negatives and false_positives should be excluded:
exclude_negatives: falsefalse_positives: false
-
Add new optional param
max_placementsto rulelinelist. -
Remove
--show-private-mutationsfromdebug_argsof rulesc2rf. -
Add optional param
--sc2rf-dirtosc2rfto enable execution outside ofsc2rfdir. -
Add params
--output-csvand--output-ansito the wrapperscripts/sc2rf.sh. -
Remove params
nextclade_refandcustom_reffrom rulenextclade. -
Change
--breakpoints 0-10insc2rf.
-
Re-rename tutorial action to pipeline, and add different jobs for different profiles:
- Tutorial
- Controls (Positive)
- Controls (Negative)
- Controls (All)
-
Output new
_historicalplots and slides for plotting all data over time. -
Output new file
parents.tsvto summarize recombinant sequences by parent. -
Order the colors/legend of the stacked bar
plotsby number of sequences. -
Include lineage and cluster id in filepaths of largest plots and tables.
-
Rename the linelist output:
linelist.tsvpositives.tsvnegatives.tsvfalse_positives.tsvlineages.tsvparents.tsv
-
The
report.xlsxnow includes the following tables:- lineages
- parents
- linelist
- positives
- negatives
- false_positives
- summary
- issues
pull/19docs: add lenaschimmel as a contributor for code
2f8b498adocs: update changelog for v0.3.00486d3bedocs: add updating section to readme for issue #33e8eda400resources: updates issues with curate breakpoints12e3700fbug: catch empty dataframe in plotd1ccca2aworkflow: first successful high-throughput runcd741a10workflow: add new rules plot_historical and report_historicalc2cc1380env: remove openpyxl from environment7dc7c039workflow: remove rule report_redact #319ca5f822script: rearrange merge file order in summaryaa28eb9fworkflow: create new rule report_redact for #314748815denv: add openpyxl to environment for excel parsing in python0060904ascript: template duplicate labelling in usher_collapse for latera82359a7data: add accession versions to controls metadataaf7341aadata: add accession versions to controls metadatad860a4c8workflow: add new rule usher_columns to augment the base usher metadata2511673dimprove subtree collapse effiency (#35) and output aesthetics (#20)1e81be3bbug: remove non-existant param --log in rule usher_metadata02198b4cscript: add logging to usher_collapsed40d3d78ci: don't run pipeline just for images changesb880d9c8docs: update powerpoint image to proper ver2d6514a0docs: update demo excel and slides with links and content59c24ffebug: fix typo that prevented low_memory_mode from activating4d94df1dbug: continue supply missing build param to params functionsc16c3377bug: supply missing build param to _params_linelistc31c2204docs: remove plotting data table from FAQ5461cbf2docs: describe how to include more custom metadata columns7295c8c0script: implement low memory mode within usher_metadata script6588f619workflow: restore original config merge logicae96cf3ddocs: rearrange sections in READMEe99cdef9docs: add tips for speeding up usher in FAQ753d1e1dresources: add proposed759 to curated breakpoints1ea5610edocs: change troubleshooting section to FAQ42152710workflow: add logging to sc2rf_recombinants for issue #34ca930fe3bug: fix status of designated recombinants missed by sc2rf (XB)2c6102a6script: in plotting data replace counts that are empty string with 00c7fa988docs: tidy up comments in default parameters.yaml43c61d43bug: fix sc2rf postprocessing bug where sequences with only parent were missing filtered regions6a00c866ci: split jobs by profile for testing profile creation (#27)aeabf009ci: add new action profile_creation to test script create_profile.sh9a6758e2add controls section to READMEef250a22script: add -controls suffix to profiles created with --controls param150a3e17docs: update troubleshooting section90b406c8script: remove --partition flag to scripts/slurm.sha0c6ece2docs: update google drive link to example slidesa37afeeadocs: update instructions for create_profile.shf9d050d2add execute permissions to scripts38b5b422bug: use a full loop to check issue formatting307b4f67catch issues list when converting to str639f8c26bug: fix issues rendering as float in tables for issue #2935ea4be1remove param --sc2rf-dir from scripts/sc2rf.sh5a2a9520docs: update images for excel and powerpoint3ae737d5env: comment out yarn which is a dev dependency3842e898improve logging in create_profile84c684caworkflow: separate profiles for controls,controls-positive,controls-negativead5e8e4blimit missing strains output from create_profilea4898ecfdocs: update development notes34ee2fffdocs: add links to contributors pluginsb6b0c999revert to automated all-contributorse1a248f8add @yatisht and @AngieHinrichs to credits for ushere3f432c4start adding contributors862757bddocs: create .all-contributorsrc [skip ci]a0532792docs: update README.md [skip ci]6e67e73fupdate unpublished regex to solve #215ba6b37bremove taxonium and chronumental from env2a5fc627add artifacts for all pipelines664b2e9bfix trailing whitespace in metadataeada2fa3fix negative controls metadata9aecd69afix plot dimensions for pptx embed657e8838fix outdir for linelist6fb389dcfix input type for controls build8ed69ce0upload tutorial pptx as artifactc6e647d2update ci profile for test action19cdb8edlint pipeline22e3aa6bsplit controls action into positives,negatives,and all33491320rename action Tutorial to Pipelineda2890d6fix profile in install action8a4d4fbblint all filesb167ea45update readme with profile creation instructions4d2848b9add script to generate new profiles and builds407f8abacheckpoint before auto-generating buildsccb3471badd new negatives dataset964a22f8(broken) script overhaul1f1ca1b4add param --sc2rf-dir to allow execution outside of main directory21541f02add exclude_negatives and exclude_false_positives to parameters0b5854a2update docs58b6396asplit controls data into positives and negatives11f9f6a4consolidate positives and negatives profiles into controls581255c8generalize hpc profiles1e2a70a4update HPC instructions in READMEa18b19e3(broken) add negatives data and profile11817639(broken) make plots and report dynamice833d151create tutorial-hpc profilec4ac5699remove redundant profile laptopc5107017remove ci profile4be07e79actually rename pipeline to tutorial89c4d6b5rename pipeline action to tutorial7614b399exclude alpha beta gamma by default from Nextcladed2c2461cupdate dev docsf9368d11remove proposed636 which is now XZ4fbd0ce4add XAA and XAB to resources65efd145add xz to resourcesf3641b19add parents slide to report and excel4e25f665add new script parents to summarize recombinants by parent0decd47acatch when no designated sequences are present189fbb2aupdate resources breakpoints3c5b4965update sc2rf with new logic for terminal deletionse37d68d9update issues and breakpoints761d41bfuse date in changelog for report3c486dbcadd zip to environment1ff37195add more info about system config
- New optional param
motifsfor rulesc2rf_recombinants. - New param
weeksfor new ruleplot. - Removed
prev_linelistparam.
- Switch from a pdf
reportto powerpoint slides for better automation. - Create summary plots.
- Split
reportrule intolinelistandreport. - Output
svgplots.
- New rule
plot. - Changed growth calculation from a comparison to the previous week to a score of sequences per day.
- Assign a
cluster_idaccording to the first sequence observed in the recombinant lineage. - Define a recombinant lineage as a group of sequences that share the same:
- Lineage assignment
- Parents
- Breakpoints or phylogenetic placement (subtree)
- For some sequences, the breakpoints are inaccurate and shifted slightly due to ambiguous bases. These sequences can be assigned to their corresponding cluster because they belong to the same subtree.
- For some lineages, global prevalence has exceeded 500 sequences (which is the subtree size used). Sequences of these lineages are split into different subtrees. However, they can be assigned to the correct cluster/lineage, because they have the same breakpoints.
- Confirmed not to use deletions define recombinants and breakpoints (differs from published)?
- Move
sc2rf_recombinants.pytopostprocess.pyin ktmeaton fork ofsc2rf. - Add false positives filtering to
sc2rf_recombinantsbased on parents and breakpoints.
- Add section
ConfigurationtoREADME.md.
c2369c75update CHANGELOG after README overhaul9c8a774eupdate autologs to exclude first blank line in notes2a8a7af5overhaul README9c2bd2f5change asterisks to dashes46d4ec81update autologs to allow more complex notes contenta01a903csplit docs into dev and todo23e8d715change color palette for plotting785b8a19add optional param motifs for sc2rf_recombinantsd1c1559erestore pptx template to regular view6adc5d32add seaborn to environment35a04471add changelog to report pptx99e98aa7add epiweeks to environment1644b1fcadd pptx report1ab93aff(broken) start plotting094530f0swithc sc2rf to a postprocess script02193d6etry generalizing sc2rf post-processing
- Fix bug in
sc2rf_recombinantsregions/breakpoints logic. - Fix bug in
sc2rfwhere a sample has no definitive substitutions.
-
Allow
--breakpoints 0-4, for XN. We'll determine the breakpoints in post-processing. -
Bump up the
min_lenofsc2rf_recombinantsto 1000 bp. -
Add param
mutation_thresholdtosc2rf. -
Reduce default
mutation_thresholdto 0.25 to catch [Issue #591](cov-lineages/pango-designation#591_. -
Bump up subtree size from 100 sequences to 500 sequences.
- Trying to future proof against XE growth (200+ sequences)
-
Discovered that
--primersinterferes with breakpoint detection, use only for debugging. -
Only use
--enable-deletionsinsc2rffor debug mode. Otherwise it changes breakpoints. -
Only use
--private-mutationstosc2rffor debug mode. Unreadable output for bulk sample processing.
-
Change
sc2rf_lineagecolumn to use NA for no lineage found.- This is to troubleshot when only one breakpoint matches a lineage.
-
Add
sc2rf_mutations_versionto summary based on a datestamp ofvirus_properties.json. -
Allow multiple issues in report.
-
Use three status categories of recombinants:
- Designated
- Proposed
- Unpublished
-
Add column
statusto recombinants. -
Add column
usher_extratousher_metadatafor 2022-05-06 tree. -
Separate out columns lineage and issue in
report. -
Add optional columns to report.
-
Fixed growth calculations in report.
-
Add a Definitions section to the markdown/pdf report.
-
Use parent order for breakpoint matching, as we see same breakpoint different parents.
-
Add the number of usher placements to the summary.
-
Set Auspice default coloring to
lineage_usherwhere possible. -
Remove nwk output from
usherandusher_subtrees:- Pull subtree sample names from json instead
-
Output
linelist.exclude.tsvof false-positive recombinants.
- Update
nextclade_datasetto 2022-04-28. - Add
taxoniumtoolsandchronumentalto environment. - Separate nextclade clades and pango lineage allele frequences in
sc2rf. - Exclude BA.3, BA.4, and BA.5 for now, as their global prevalence is low and they are descendants of BA.2.
-
Add a
tutorialprofile.- (N=2) Designated Recombinants (pango-designation)
- (N=2) Proposed Recombinants (issues, UCSC)
- (N=2) Unpublished Recombinants
-
Add XL to
controls. -
Add XN to
controls. -
Add XR to
controls. -
Add XP to
controls.
-
Split
usher_subtreeandusher_subtree_collapseinto separate rules.- This speeds up testing for collapsing trees and styling the Auspice JSON.
-
Force include
Nextcladerecombinants (auto-pass throughsc2rf). -
Split
usherandusher_statsinto separate rules.
pull/13Three status categories: designated, proposed, unpublished
10388a6eupdate docs for v0.2.032b9e8abseparate usher and usher_stats rule and catch 3 or 4 col usher70da837cupdate github issues and breakpoints for 636 and 6379ed10f17skip parsing github issues that don't have body216cb28eput the date in the usher data for tutorialc95cca0eupdate usher v0.5.398c91beefinish reporting cycle 2022-05-11e4755f16new sc2rf mutation data by clade4a501d56separate omicron lineages from omicron cladesd6185aaftesting change to auto-pass nextclade recombinants2e02922fadd XL XN XR XP to controlse2c9675badd usher_extra and qc file to sc2rf recombinants941a64c5update github issues943cde95add usher placements to summary0d0ffbd4combine show-private-mutations with ignore-sharedf1d7e6c1update sc2rf after terminal bugfixes8f4fd95aadd country England to geo resefeeb6caadd mutation threshold param sep for sc2rf9c42bc6climit table col width size in report7d746e3ffix growth calculation0dc7f464identify sc2rf lineage by breakpoints and parents19fc3721add parents to breakpoints and issues27bbff0agenerate geo_resolutions from ncov defaults lat longs86aa78baadd map to auspice subtrees2499827eadd taxoniumtools and chronumental to envec46a569change tutorial seq names from underscores to dashes27170a0bfix issues line endingse8eb1215update nextclade dataset to 2022-04-2889335c3a(broken) updating columns in report67475eccupdate sc2rf79b7b2b9add tip to readme10df6a54remove all sample extraction from usher2ffbcb61switch sc2rf submodule to ktmeaton forke2adaabedisable snakemake report in pipeline cif12fef14edit line linst preview instructionsd10eb730add collection date to tutoriald4e0aa86very preliminary credits and tutoriale9c41e6echange ci pipeline to tutorial build4de7370dadd tutorial data8d8c88fcset min version for click to troubleshoot env creationc7fb50a4better issue reportingb2699823update sc2rf
-
Add lineage
XMto controls.- There are now publicly available samples.
-
Correct
XFandXJcontrols to match issues. -
Create a markdown report with program versions.
-
Fix
sc2rf_recombinantsbug where samples with >2 breakpoints were being excluded. -
Summarize recombinants by parents and dates observed.
-
Change
report.tsvtolinelist.tsv. -
Use
date_to_decimal.pyto createnum_datefor auspice subtrees. -
Add an
--exclude-cladesparam tosc2rf_recombinants.py. -
Add param
--ignore-shared-substosc2rf.- This makes regions detection more conservative.
- The result is that regions/clade will be smaller and breakpoints larger.
- These breakpoints more closely match pango-designation issues.
-
Update breakpoints in controls metadata to reflect the output with
--ignore-shared-subs. -
Bump up
min_lenforsc2rf_recombinantsto 200 bp. -
Add column
sc2rf_lineagetosc2rf_recombinantsoutput.- Takes the form of X*, or proposed{issue} to follow UShER.
-
Consolidate lineage assignments into a single column.
- sc2rf takes priority if a single lineage is identified.
- usher is next, to resolve ties or if sc2rf had no lineage.
-
Slim down the conda environment and remove unnecessary programs.
augurseabornsnipitbedtools- Comment out dev tools:
gitandpre-commit.
-
Use github api to pull recombinant issues.
-
Consolidate *to* files into
resources/issues.tsv. -
Use the
--cladesparam ofsc2rfrather than usingexclude_clades. -
Disabled
--rebuild-examplesinsc2rfbecause of requests error. -
Add column
issuetorecombinants.tsv. -
Get
ncov-recombinantversion using tag. -
Add documentation to the report.
- What the sequences column format means: X (+X)
- What the different lineage classifers.
pull/10Automated report generation and sc2rf lineage assignments
941f0c08update CHANGELOGbce219b6fix notes conflictcdb3bc7ffix duplicate pr output in autologs0a8ffd84update notes for v0.1.20075209badd issue to recombinants report0fd8fea0(broken) troubleshoot usher collapse jsonb0ab72b9update breakpointsa3058c57update sc2rf examplesb5f2a0f8update and clean controls dataf69f254cadd curated breakpoints to issues4e9d2dc9add an issues script and resources file16059610major environment reduction5ced9193remove bedtools from env28777cb9remove seaborn from env01f459c4remove snipit from env7d6ef729remove augur from env9a2e948dfix pandas warnings in report78daee55remove script usher_plot as now we rely on auspicea7bd52f1remove script update_controls which is now done manually93a30200consolidate lineage call in report5f3e3633hardcode columns for report6333fde4improve type catching in date_to_decimal557627a4update controls breakpoints and add col sc2rf_lineage7e2ad531add param --ignore-shared-subs to sc2rf519a9eeaoverhaul reporting workflow6fa674f2update controls metadata and dev notes46155a84add XM to controlsd2d4cd80update notes for v0.1.217ae6eebadd issue to recombinants report2ab8dd30(broken) troubleshoot usher collapse jsone4fd3352update breakpointsc743ad3dupdate sc2rf examples62e1ffc1update and clean controls data9c401a0cadd curated breakpoints to issues7cf953adadd an issues script and resources filefbf35e51major environment reduction1b1f16e9remove bedtools from env7b650279remove seaborn from envb32608e7remove snipit from env9cab231eremove augur from enva69836a8fix pandas warnings in reportb3137ed3remove script usher_plot as now we rely on auspice06fa080dremove script update_controls which is now done manuallye8d46a64consolidate lineage call in reportccfea688hardcode columns for report5172755eimprove type catching in date_to_decimal74f0e528update controls breakpoints and add col sc2rf_lineaged4664015add param --ignore-shared-subs to sc2rf659d7f83overhaul reporting workflowe8e3444fupdate controls metadata and dev notes07a5ff52add XM to controls
-
Add lineage
XDto controls.- There are now publicly available samples.
-
Add lineage
XQto controls.- Has only 1 diagnostic substitution: 2832.
-
Add lineage
XSto controls. -
Exclude lineage
XRbecause it has no public genomes.XRdecends fromXQin the UShER tree.
-
Test
sc2rfdev to ignore clade regions that are ambiguous. -
Add column
usher_pango_lineage_mapthat maps github issues to recombinant lineages. -
Add new rule
report. -
Filter non-recombinants from
sc2rfansi output. -
Fix
subtrees_collapsefailing if only 1 tree specified -
Add new rule
usher_metadatafor merge metadata for subtrees.
b8f89d5eupdate docs for v0.1.12b8772abupdate autologs for pr date matchingf2a7547dadd low_memory_mode for issue #994fc9426add log to report rule861ffb17update usher output imagefdee0da6add new rule usher_metadatac5003453add max parents param to sc2rf recombinants70cad049add max ambig filter to defaults for sc2rfb4cc40f4add script to collapse usher metadata for auspice847c6d24catch single trees in usher collapse636778e0rename sc2rf txt output to ansibae50814change final target to report9a830085add new rule report601e1728add XD to controlsbaa1d64erelax sc2rf --unique from 2 to 1 for XQbffbb9adadd column sc2rf_clades_filter109ed5d2test sc2rf dev to not report ambiguous regionsd9fdffeffix tab spaces at end of usher placement085e1764update sc2rf for tsv/csv PRd2363855set threads and cpus to 1 for all single-thread rules13027205impose wildcard constraint on nextclade_dataset30e1406bfix typo in csv path for sc2rfd6d8377aadd XS breakpoints to metadata371e4069add XS and XQ to controls1e31e429add breakpoints reference file in controls3088ad60catch if sc2rf has no output64211360catch all extra args in slurmc7a6b9ceremove unused nextclade_recombinants script38645ce9remove codecov badge50fa9d89update CHANGELOG for v0.1.0
-
Add Stage 1: Nextclade.
-
Add Stage 2: sc2rf.
-
Add Stage 3: UShER.
-
Add Stage 4: Summary.
-
Add Continuous Integration workflows:
lint,test,pipeline, andrelease. -
New representative controls dataset:
- Exclude XA because this is an Alpha recombinant (poor lineage accuracy).
- Exclude XB because of current issue
- Exclude XC because this is an Alpha recombinant (poor lineage accuracy).
- Exclude XD because there are no public genomes.
- Exclude XK because there are no public genomes.
pull/3Add Continuous Integration workflows: lint, test, pipeline, and release.
34c721b7rearrange summary cols18b389dedisable usher plotting0a101dd9covert sc2rf_recombinants to a script494fb60cchange nextclade filtering to multi columnsf0676bd8add python3 directive to unit tests9da626fdupdate unit tests for new controls dataset46717c6ffix summary script for new ver9fc690edfix usher public-latest links1fb28ea0change sc2rf to bash scriptba98d936update sc2rf params89647109update sc2rf artpoon pr2b7dcab4ignore my_profiles9df0f9b4add debugging mode for sc2rf (lint)caaf4debadd debugging mode for sc2rf7ec53ac6remove unecessary param exp_inputa3810bcaadd program versions to summary3376196bAdd Continuous Integration workflows: lint, test, pipeline, and release. (#3)db45768fmore instructions for visualizing outputee4c2660update readme keywordscb5b2c23add instructions for slurm submissionf04eb28badjust laptop profile to use 1 cpueafed44eadd snakefileeb214d72add scriptscb47b046add README.md3794d8deadd images dirf1c0cef8add profiles laptop and hpc6f5fd84badd release notes92b0c476add default parametersc79bfe12add submodule ncov2a65e92dadd submodule sc2rf0f8bfe33add submodule autologsb6f1e1d6add report captions68fd2decadd conda env8907375eadd reference dataset