
![]()

A snakemake wrapper and utility tools for BGCFlow, a systematic workflow for the analysis of biosynthetic gene clusters across large genomic datasets.
For more details, see documentation.
Please refer to the BGCFlow WIKI for detailed examples and use cases:
Matin Nuhamunada, Omkar S Mohite, Patrick V Phaneuf, Bernhard O Palsson, Tilmann Weber, BGCFlow: systematic pangenome workflow for the analysis of biosynthetic gene clusters across large genomic datasets, Nucleic Acids Research, 2024;, gkae314, https://doi.org/10.1093/nar/gkae314
To install bgcflow_wrapper with conda/mamba, run this command in your
terminal:
# create and activate new conda environment
mamba create -n bgcflow -c conda-forge python=3.11 pip openjdk -y
conda activate bgcflow
# install BGCFlow wrapper
pip install bgcflow_wrapper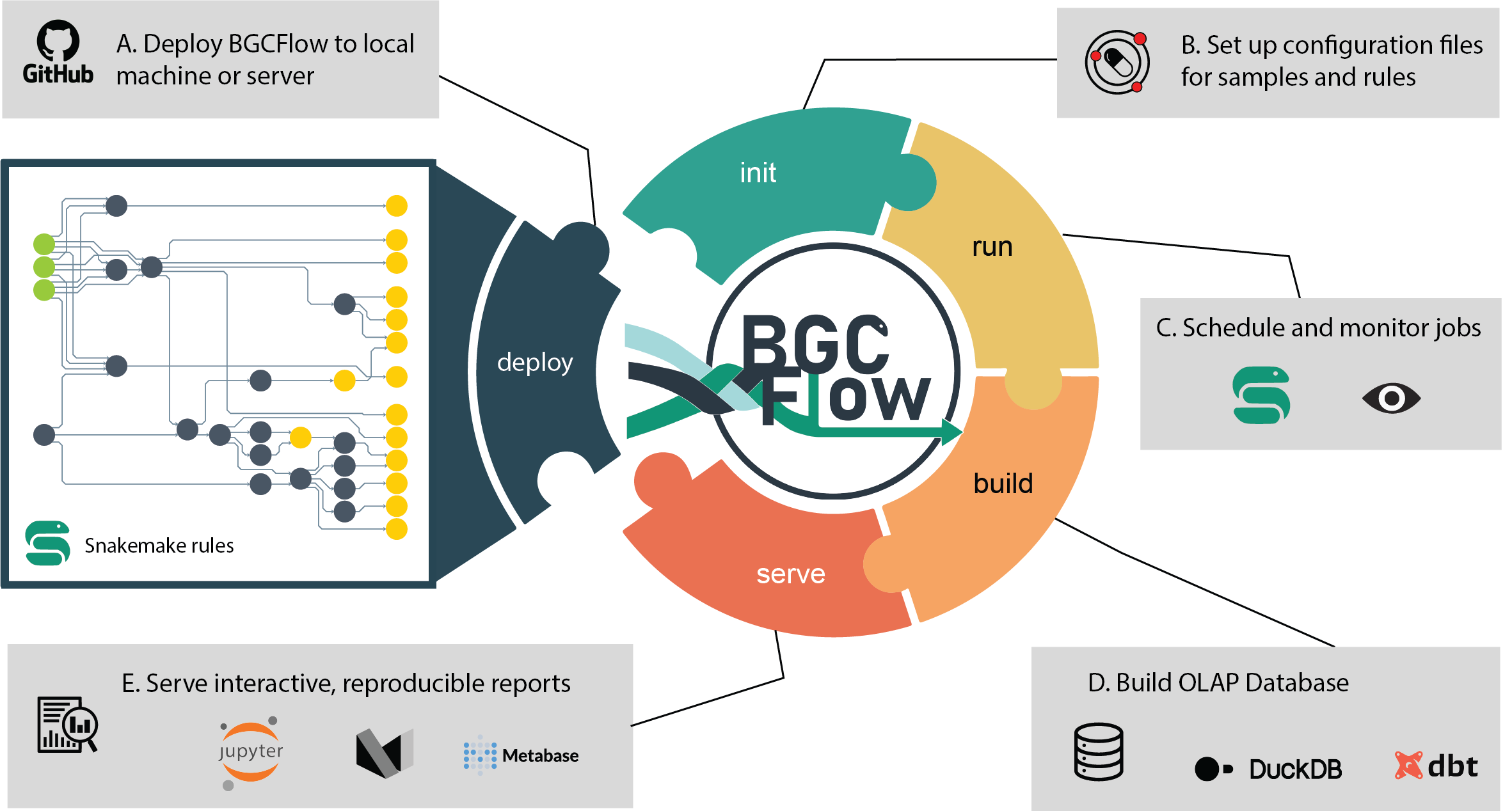
$ bgcflow
Usage: bgcflow [OPTIONS] COMMAND [ARGS]...
A snakemake wrapper and utility tools for BGCFlow
(https://github.com/NBChub/bgcflow)
Options:
--version Show the version and exit.
-h, --help Show this message and exit.
Commands:
build Build Markdown report or use dbt to build DuckDB database.
clone Get a clone of BGCFlow to local directory.
deploy [EXPERIMENTAL] Deploy BGCFlow locally using snakedeploy.
get-result View a tree of a project results or get a copy using Rsync.
init Create projects or initiate BGCFlow config from template.
pipelines Get description of available pipelines from BGCFlow.
run A snakemake CLI wrapper to run BGCFlow.
serve Serve static HTML report or other utilities (Metabase, etc.).
sync Upload and sync DuckDB database to Metabase.This package was created with the ppw tool. For more information, please visit the project page.