{kind=link}
Anczukow-lab repository for the Nextflow splicing pipeline with rMATS 4.1.0 (tag = v1.0) and rMATS 4.1.1 (master).
The workflow processes raw data from FastQ inputs (FastQC, Trimmomatic), aligns the reads (STAR), performs transcript assembly and quantification (StringTie), detects alternative splicing events (rMATs), and generates an interactive QC report (MultiQC).
The pipeline is built using Nextflow, a workflow tool to run tasks across multiple compute infrastructures in a very portable manner. For example, it can be run on HPC (using JAX's Sumner HPC) or on the cloud over (Lifebit's CloudOS platform with AWS & GCloud). It comes with docker containers making installation trivial and results highly reproducible.
The input reads to this pipeline can come from 3 input sources:
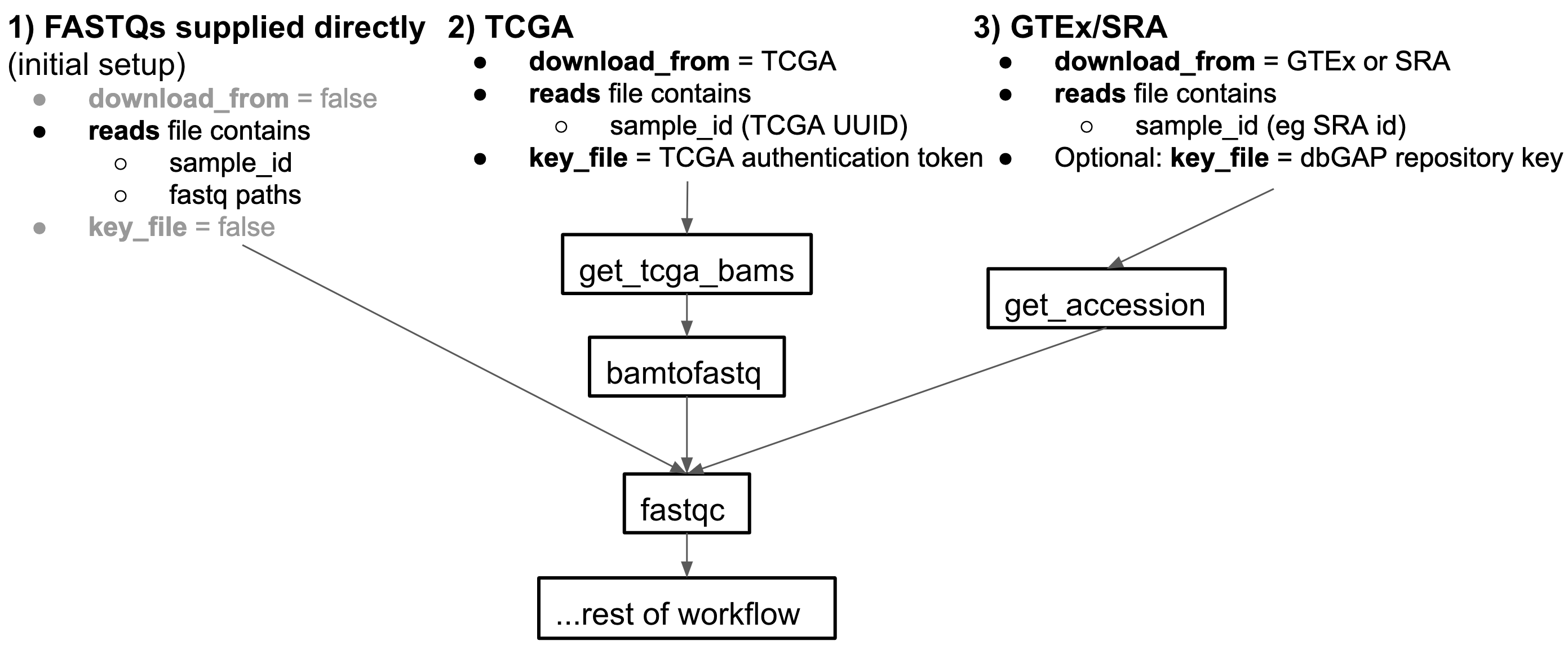
- Go into splicing pipeline folder
cd /projects/anczukow-lab/splicing_pipeline/splicing-pipelines-nfstatusto check branch and if there are unmerged changes. Note:masteris the main branch.
git statuspullany new changes. Note: you will need your github username and password
git pull-
Create new directory to run test and
cdinto that folder. Make this folder in individual's folder withinanczukow-lab -
Run test:
sbatch /projects/anczukow-lab/splicing_pipeline/splicing-pipelines-nf/examples/human_test/human_test_main.pbs- To check progress:
squeue -u [username]or by looking at tail of output file:
tail splicing.[jobnumber].outnextflow run main.nf --help
Documentation about the pipeline, found in the docs/ directory:
- Intros to
- Pipeline overview
- Running the pipeline

View changelog at changelog.md