The Pan-Genome Analysis Pipeline
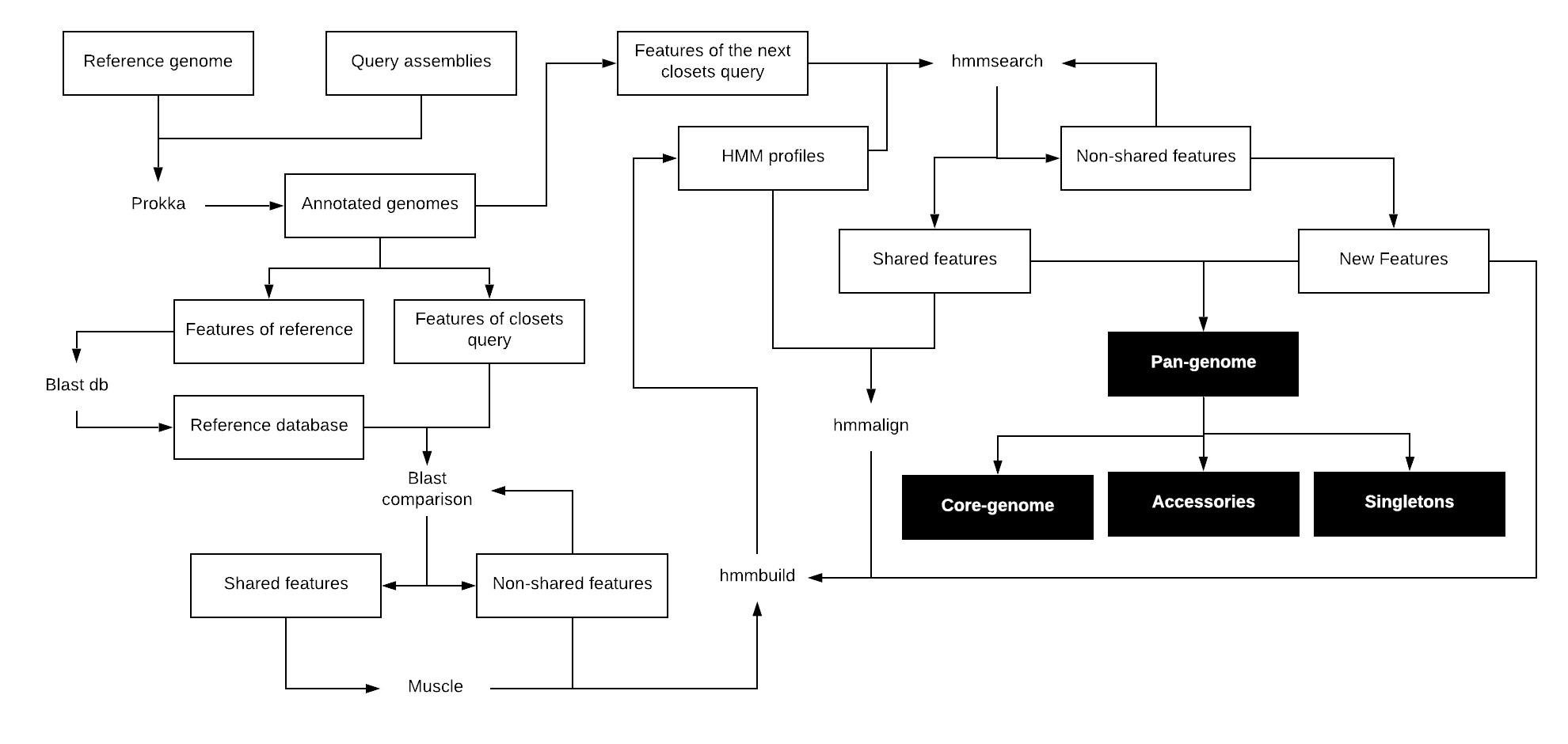
Pangea is a pipeline to calculate the entire gene collection of a set of prokaryotic organisms (Pan genome). It is based on an all-against-all algorithm to find shared and non-shared genes between samples.
Pangea was designed to compare high diverse prokaryotic genomes using profile Hidden Markov Model (profile HMM) to make a position-specific scoring. Pangea builds a profile HMM for each gene that constitutes the pan genome. Then each profile is used to search the corresponding gene in each sample. During this search the profiles are iteratively populated with the corresponding homologous sequences. This allows each profile HMM to include all the variations that a gene presents between samples, increasing the robustness and therefore the accuracy of the search.
First, Pangea makes a single blast comparison (blastn for nucleotides or blastp for amino acids) between the features of the first two query samples or between the first query and a reference sample. Homologous sequences, according to an established e-value and a percentage of identity, are considered shared features, while features without any blast hit are considered non-shared. Sequences of shared features are extracted and aligned with muscle to build a profile HMM from the alignment using hmmbuil, while sequences of shared features are extracted and used to build profile HMM from a single sequence. Then, the profiles HMM are used to look for homologous features in the remaining samples according to the same percentage of identity and e-value used in the blast comparison. Only the best hit is considered homolog (shared feature) and its sequence is added to the corresponding profile HMM, while non-homologous features are re-analyzed to discard duplicates or fragmented sequences, which would generate less significant hits. Remaining non-homologous features are considered new elements of the pan genome and their sequence are used to create profiles HMM from a single sequence. All features are reported in a presence/absence table, which represents the composition of the entire pan genome, including the core genome, accessory genome and singletons.
Pangea is a collection of perl scripts and it has been tested on Linux and MacOS. For its correct execution requires the previous installation of some third-party programs.
For now, Pangea works only with the outputs produced by Prokka. All the annotated samples should be contained in the same directory, one folder per sample. It is highly recommended to use the --locustag option during the annotation process with prokka. For more information on the installation and execution of prokka check here.
You have to create a text file that lists all the samples you want to analyze. The samples will be analyzed according to the order specified in the list.
Additionally you can include a reference sample. This can be declared at the beginning of your sample list or you can use the --trusted option to include it. See the Usage section.
Depending on the used options, the possible outputs include the following:
| Name | Type | Description |
|---|---|---|
| Blast | Dir | Contains all the blast databases used by the program |
| PanGenomeHmmDb | Dir | Contains a database of the pan genome used during the HMMER comparisons |
| ORFeomes | Dir | Contains the ORFeomes or Proteomes of the queries |
| ORFs | Dir | Contains all the analyzed features. One folder per feature |
| CoreSequences | Dir | Contains the core and the aligned-core fasta files of each query |
| *UniqueBlastComparison.txt | File | Contains the blast report of the first comparison |
| *Progress.csv | File | Records the progress of the analysis. It includes the total number of features, the size of the pan-genome and the number of new genes added in each sample analyzed |
| *Summary.txt | File | Contains a summary of the parameters used for the analysis and the final size of the pan and core genome |
| *Presence_Absence.csv | File | Contains the record of the features present in each sample. Each gene is identified by its locus tag |
| *CoreGenome.csv | File | Contains the record of the features present in the core genome defined as features present in the 100% of samples. Each gene is identified by its locus tag |
| *Boolean_PanGenome.csv | File | Contains the record of the features present in each sample. Presence of a feature is represented by 1 while absence is represented by 0 |
| *Boolean_AccessoryGenes.csv | File | Contains the record of the dispensable features defined as features present in more than one sample but not present in the 100% of samples. Presence of a feature is represented by 1 while absence is represented by 0 |
| *PanGenome.fasta | File | Multifasta file with the sequences of features of the final pan genome. Features included were extracted from the first sample that each feature was detected |
| *ConsensusPanGenome | File | Multifasta file with the consensus sequence of the pan genome obtained from the profile HMM of each feature |
| *PanGenome_HeatMap.pdf | Plot | Representation of the Presence/Absence of fueatures of the pan genome |
Usage:
$./Pangea.pl [options] --list Full/Path/Of/Sample/List --annotation /Full/Path/Of/Annotation/Directory
--moltype nucl/prot --out Full/Path/For/Outputs
Options:
--help Print this help message
--project -p STR Set a prefix name for the project
--list -l STR File with the list of samples. Full path is mandatory
--trusted -c STR Name of the sample that you want to use as reference.
Annotation of reference shuld to be included in the same
directory as the rest of the samples
--evalue -e REAL Expectation value threshold used on blast and HMMER comparisons
--ident -i REAL Minimum percentage of identity for blast and HMMER
--cpus -t INT Number of threads used only on some steps of blast and HMMER searches
--conpan -P Create a multifasta file of consensus pan genes
--alncore -C Creaate a multifasta file of aligned core genes
--booleantbl -b Create gene presence/absence table for the pan and accessory genes
where presence is represented by 1 and abence by 0
--annotation -a STR Full path of the annotation directory
--moltype -m STR Type of molecule you want to analyze, "nucl" for nucleotides
and "prot" for aminoacids
--out -o STR Output directory
--recovery -r Recover an interrupted process and/or increase the analysis
with additional samples. Increase the analysis requires adding
the new samples to the sample list
Seemann T.
Prokka: rapid prokaryotic genome annotation
Bioinformatics 2014 Jul 15;30(14):2068-9.
PMID:24642063
Website: https://torresrc.github.io/Pangea/