This repository contains the workflow of RNAseq data analysis.
This pipeline performs the following tasks:
- Reading data
- align reads of each sample in a run against reference genome = perform quality control on generated BAM files
- count reads in features
- normalize read counts
- Filtering lowly expressed genes
- perform DE analysis
The data used for analysis is from the study, “EGF-mediated induction of Mcl-1 at the switch to lactation is essential for alveolar cell survival” (Fu et al. 2015).
This study examines the expression profiles of basal stem-cell enriched cells and committed luminal cells in the mammary gland of virgin, pregnant and lactating mice.
GEO Accession ID - GSE60450
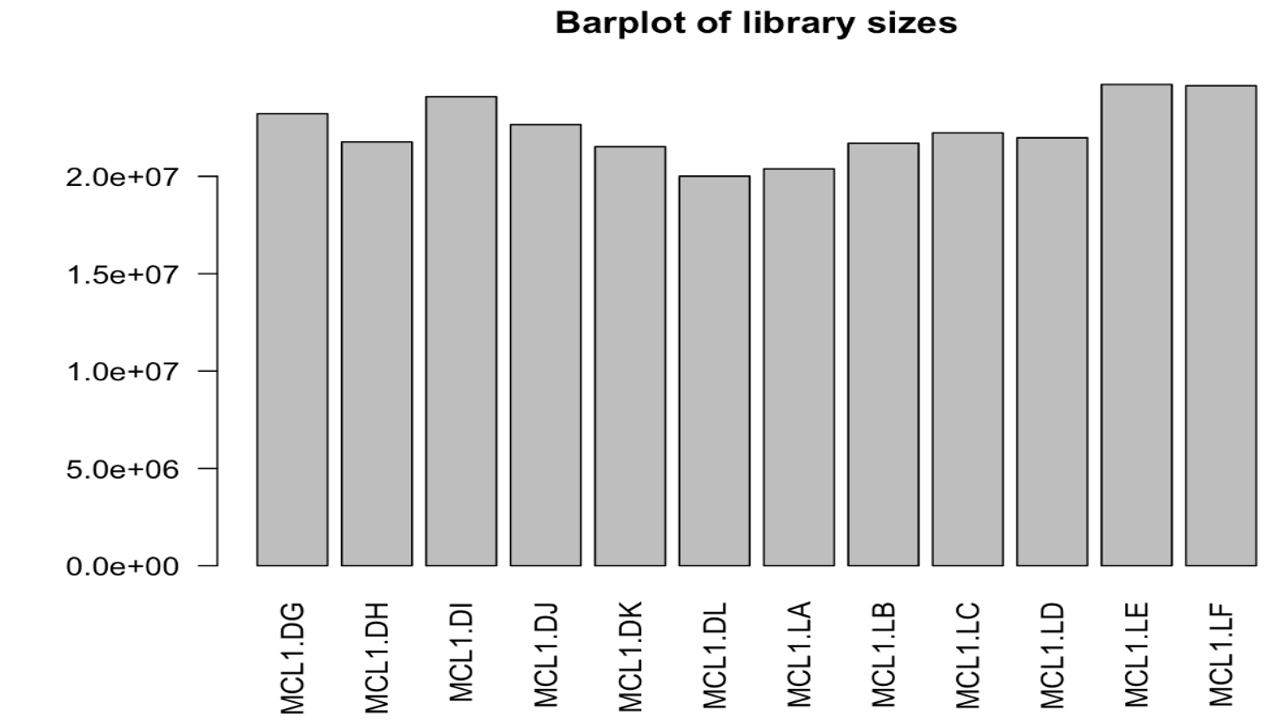
The read count data, metadata and the fastq files are present in Datasets folder.
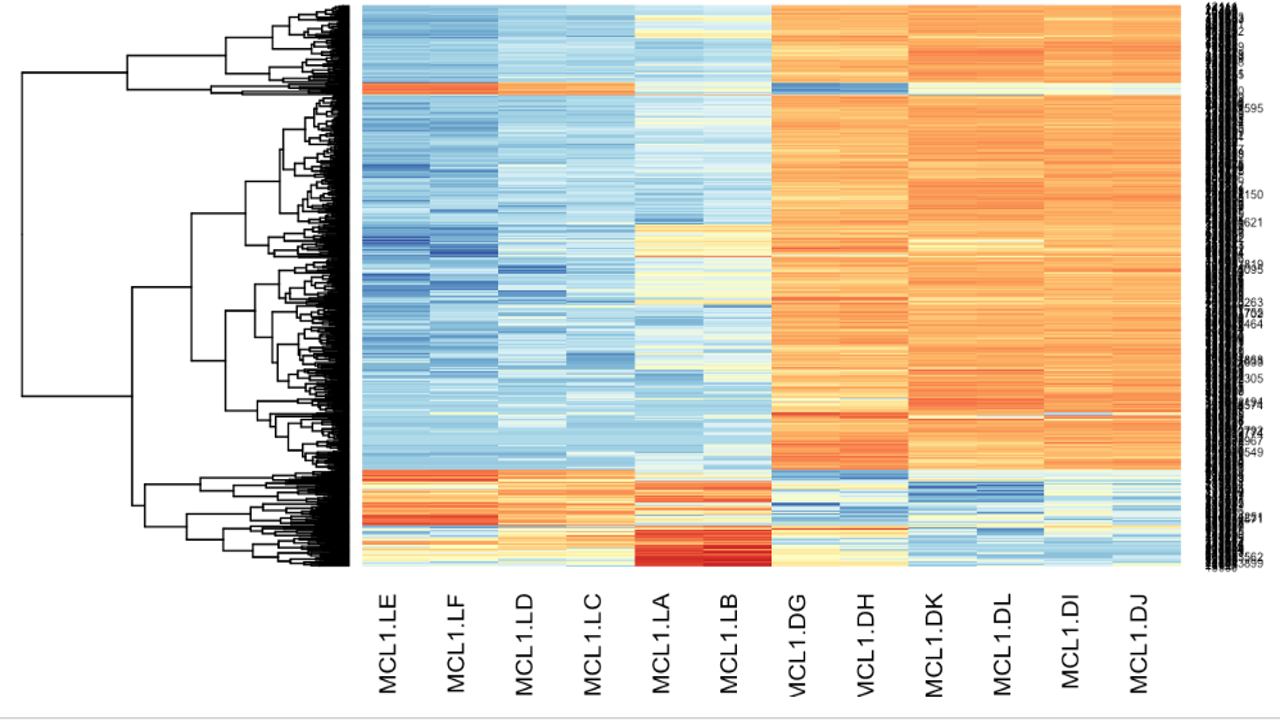
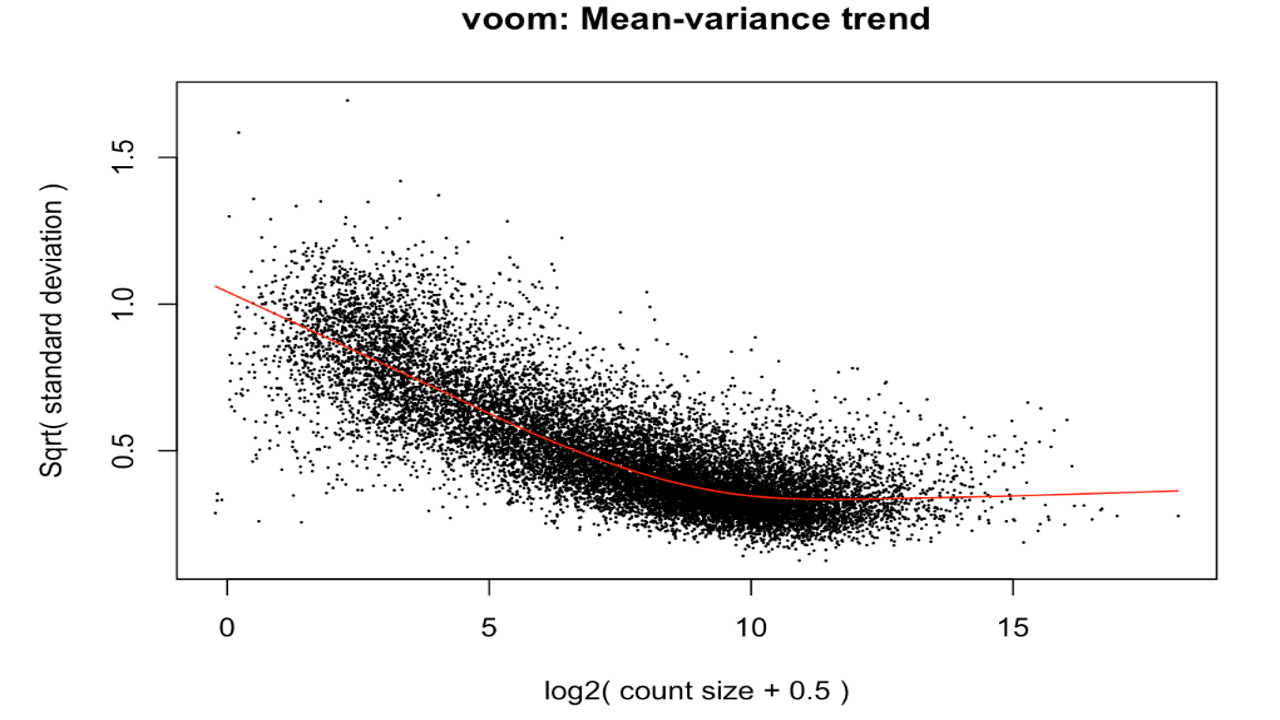
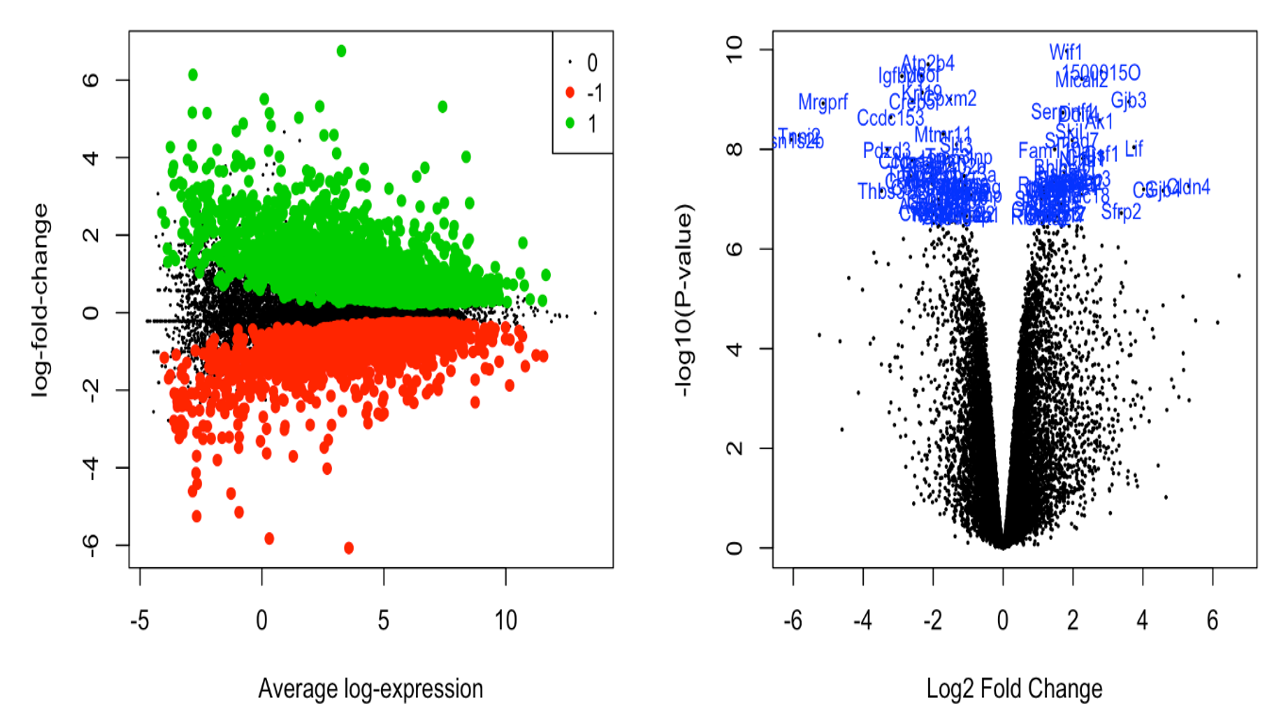
The differentially expressed genes were obtained. Various plots like heatmap, mean-variance plot, MA plot, and volcano plot were made to analyse the data better.