


MDBenchmark — quickly generate, start and analyze benchmarks for your molecular dynamics simulations.
MDBenchmark is a tool to squeeze the maximum out of your limited computing resources. It tries to make it as easy as possible to set up systems on varying numbers of nodes and compare their performances to each other.
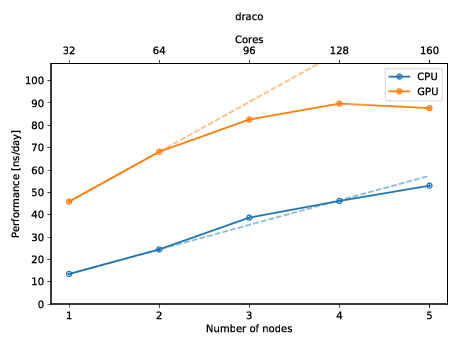
You can also create a plot to get a quick overview of the possible performance (and also show of to your friends)! The plot below shows the performance of a molecular dynamics system on up to five nodes with and without GPUs.
You can install mdbenchmark via pip, conda or pipenv:
pip install mdbenchmark
conda install -c conda-forge mdbenchmark
pipx install mdbenchmark
pipenv install mdbenchmark
After installation MDBenchmark is accessible on your command-line via mdbenchmark:
$ mdbenchmark Usage: mdbenchmark [OPTIONS] COMMAND [ARGS]... Generate, run and analyze benchmarks of molecular dynamics simulations. Options: --version Show the version and exit. --help Show this message and exit. Commands: analyze Analyze benchmarks and print the performance... generate Generate benchmarks for molecular dynamics... plot Generate plots showing the benchmark... submit Submit benchmarks to queuing system.
- Generates benchmarks for GROMACS and NAMD simulations (contributions for other MD engines are welcome!).
- Automatically detects the queuing system on your high-performance cluster and submits jobs accordingly.
- Grabs performance from the output logs and creates a fancy plot.
- Benchmarks systems on CPUs and/or GPUs.
- Find the best parameters by scanning different numbers of MPI ranks and OpenMP threads.
- Run multiple instances of the same simulation on a single node using GROMACS'
--multidiroption.
The following shows a short usage reference for MDBenchmark. Please consult the documentation for a complete guide.
Assuming you want to benchmark GROMACS version 2018.3 and your TPR file is
called protein.tpr, run the following command:
mdbenchmark generate --name protein --module gromacs/2018.3
To run benchmarks on GPUs simply add the --gpu flag:
mdbenchmark generate --name protein --module gromacs/2018.3 --gpu
After you generated your benchmarks, you can submit them at once:
mdbenchmark submit
As soon as the benchmarks have been submitted you can run the analysis via
mdbenchmark analyze. Systems that have not finished yet will be marked with a question mark (?). You can save the performance results to a CSV file and subsequently create a plot from the data:
# Print performance results to console and save them to a file called results.csv mdbenchmark analyze --save-csv results.csv # Create a plot from the results present in the file results.csv mdbenchmark plot --csv results.csv
Please cite the latest MDBenchmark publication if you use the tool to benchmark your simulations. This will help raise awareness of benchmarking and help people improve their simulation performance, as well as reduce overall resource wastage.
M. Gecht, M. Siggel, M. Linke, G. Hummer, J. Köfinger MDBenchmark: A toolkit to optimize the performance of molecular dynamics simulations. J. Chem. Phys. 153, 144105 (2020); https://doi.org/10.1063/5.0019045
Contributions to the project are welcome! Information on how to contribute to the project can be found in CONTRIBUTING.md and DEVELOPER.rst.