Genome-wide detection of human intronic AG-gain variants between the splicing branchpoint and acceptor site
-
Human variants that introduce an AG in the intronic region between the branchpoint (BP) and the canonical splice acceptor site (ACC) of protein-coding genes can disrupt pre-mRNA splicing.
-
Based on our previously developed BPHunter (GitHub and webserver), we precisely delineate the BP-ACC segments of all human introns of protein-coding genes, for the first time. We find an extreme depletion of AG/YAG in the high-risk [BP+8, ACC-4] region.
-
AGAIN is a genome-wide approach to pinpoint intronic AG-gain variants (SNVs, insertions, deletions) in the BP-ACC region of human genomes. AGAIN also predicts protein-level outcomes resulting from two major missplicing consequences (new acceptor site & complete exon skipping).
-
This standalone version can be implemented into NGS analysis by a one-line command. We also provide the AGAIN webserver with a user-friendly interface.
- Feb 2023: AGAIN webserver & GitHub was launched.
- Sep 2022: AGAIN prototype was completed.
- Jan 2022: Inspired by the BPHunter study, AGAIN idea was conceived.
Current version: version-1
The code is written in python3, and requires bedtools installed.
Due to the data limit in GitHub, please download the AGAIN reference datasets and put them into your AGAIN folder.
Input: Variants in VCF format, with 5 mandatory and tab-delimited fields (CHROM, POS, ID, REF, ALT), (Sample_variants.vcf)
Output: AGAIN-detected variants will be output with the following annotations.
- SAMPLE (only for AGAIN_VCF_batch.py)
- CHROM, POS, ID, REF, ALT (exactly the same as input)
- STRAND: +/-
- VAR_TYPE: snv, x-nt del, x-nt ins
- GENE: gene symbol
- TRANSCRIPT_IVS: ENST123456789_IVS10
- CANONICAL: canonical transcript_IVS, or '.'
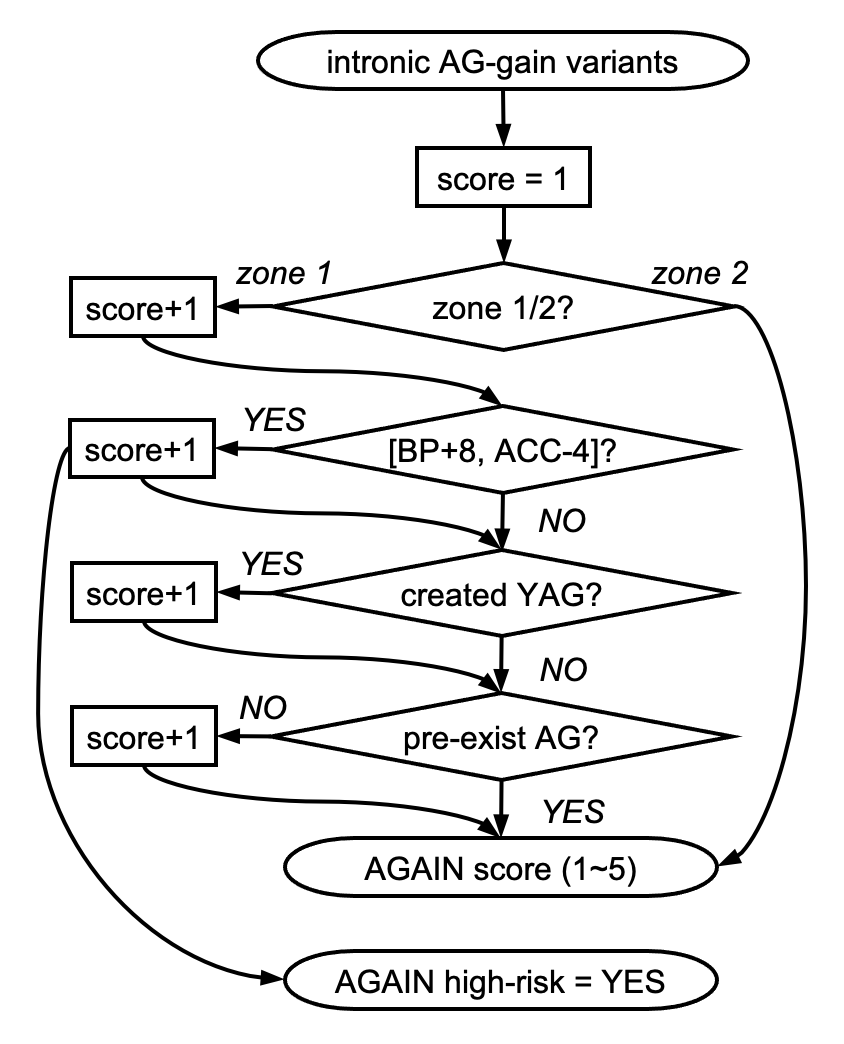
- AGAIN_ZONE: ZONE1/ZONE2, ZONE1 is from 1st BP to ACC, ZONE2 is from 2nd BP to 1st BP
- AGAIN_YAG: YES/NO, if the AG-gain variant also fits YAG
- AGAIN_BP_DIST: the distance from the created AG to BP
- AGAIN_ACC_DIST: the distance from the created AG to ACC
- AGAIN_HIGHRISK: YES/NO, if the AG-gain variant falls inside the high-risk [BP+8, ACC-4] region
- AGAIN_SCORE: score of the AG-gain variant (suggested cutoff >= 3, max = 5)
- PROT_SEQ_WT: wild-type protein sequence
- PROT_SEQ_NEW_ACC: consequent protein sequence if a new acceptor site is created
- HGVS_NEW_ACC: protein-level HGVS annotation if a new acceptor site is created
- PROT_SEQ_EXON_SKIP: consequent protein sequence if exon skipping occurs
- HGVS_EXON_SKIP: protein-level HGVS annotation if exon skipping occurs
python AGAIN_VCF.py -i Sample_variants.vcf
python AGAIN_VCF.py -g GRCh37 -t all -i Sample_variants.vcf
| Parameter | Type | Description | Default |
|---|---|---|---|
| -g | str | human reference genome assembly (GRCh37 / GRCh38) | GRCh37 |
| -t | str | all / canonical transcripts? | all |
| -i | file | variants in VCF format, with 5 fields (CHROM, POS, ID, REF, ALT) | N.A. |
python AGAIN_VCF_batch.py -d directory -s samplelist.txt -o output.csv
python AGAIN_VCF_batch.py -g GRCh37 -t all -d directory -s samplelist.txt -o output.csv
| Parameter | Type | Description | Default |
|---|---|---|---|
| -g | str | human reference genome assembly (GRCh37 / GRCh38) | GRCh37 |
| -t | str | all / canonical transcripts? | all |
| -d | str | directory of VCF files | N.A. |
| -s | file | sample list (without .vcf extension) in the above directory | N.A. |
| -o | str | output filename in csv | N.A. |
- Zhang P. et al. Genome-wide detection of human intronic AG-gain variants located between the splicing branchpoint and canonical splice acceptor site. 2023.
Developer: Peng Zhang, Ph.D.
Email: pzhang@rockefeller.edu
Laboratory: St. Giles Laboratory of Human Genetics of Infectious Diseases
Institution: The Rockefeller University, New York, NY, USA