




Contents
ChemPy is a Python package useful for chemistry (mainly physical/inorganic/analytical chemistry). Currently it includes:
- Numerical integration routines for chemical kinetics (ODE solver front-end)
- Integrated rate expressions (and convenience fitting routines)
- Solver for equilibria (including multiphase systems)
- Relations in physical chemistry:
- Debye-Hückel expressions
- Arrhenius & Eyring equation
- Einstein-Smoluchowski equation
- Properties (pure python implementations from the literature)
- water density as function of temperature
- water permittivity as function of temperature and pressure
- water diffusivity as function of temperature
- water viscosity as function of temperature
- sulfuric acid density as function of temperature & weight fraction H₂SO₄
- More to come... (and contributions are most welcome!)
The easiest way to get started is to have a look at the examples in this README, and also the jupyter notebooks. In addition there is auto-generated API documentation for the latest stable release here (and here are the API docs for the development version).
Simplest way to install ChemPy and its (optional) dependencies is to use the conda package manager:
$ conda install -c bjodah chempy pytest $ pytest -rs -W ignore::chempy.ChemPyDeprecationWarning --pyargs chempy
currently conda packages are only provided for Linux. On Windows and OS X
you will need to use pip instead:
$ python3 -m pip install chempy pytest $ python3 -m pytest -rs -W ignore::chempy.ChemPyDeprecationWarning --pyargs chempy
there will a few tests which will be skipped due to some missing optional backends in addition to those in SciPy (used for solving systems of non-linear equations and ordinary differential equations).
If you are still using Python 2 you can use the long-term-support 0.6.x branch of ChemPy which will continue to receive bugfixes:
$ python2 -m pip install "chempy<0.7"
If you used conda to install ChemPy you can skip this section.
But if you use pip the default installation is achieved by writing:
$ python3 -m pip install --user --upgrade chempy pytest $ python3 -m pytest -rs --pyargs chempy
you can skip the --user flag if you have got root permissions.
You may be interested in using additional backends (in addition to those provided by SciPy)
for solving ODE-systems and non-linear optimization problems:
$ python3 -m pip install chempy[all]
Note that this option will install the following libraries (some of which require additional libraries to be present on your system):
- pygslodeiv2: solving initial value problems, requires GSL. (>=1.16).
- pyodeint: solving initial value problems, requires boost (>=1.65.0).
- pycvodes: solving initial value problems, requires SUNDIALS (>=5.3.0).
- pykinsol: solving non-linear root-finding, requires SUNDIALS (>=5.3.0).
- pycompilation: python front-end for calling compilers, requires gcc/clang/icpc.
- pycodeexport: package for code-generation, used when generating C++ code.
if you want to see what packages need to be installed on a Debian based system you may look at this Dockerfile.
If you have Docker installed, you may use it to host a jupyter notebook server:
$ ./scripts/host-jupyter-using-docker.sh . 8888
the first time you run the command, some dependencies will be downloaded. When the installation is complete there will be a link visible which you can open in your browser. You can also run the test suite using the same docker-image:
$ ./scripts/host-jupyter-using-docker.sh . 0
there will be a few skipped test (due to some dependencies not being installed by default) and quite a few warnings.
See demonstration scripts in examples/, and some rendered jupyter notebooks. You may also browse the documentation for more examples. Below you will find a few code snippets:
>>> from chempy import Substance
>>> ferricyanide = Substance.from_formula('Fe(CN)6-3')
>>> ferricyanide.composition == {0: -3, 26: 1, 6: 6, 7: 6} # 0 for charge
True
>>> print(ferricyanide.unicode_name)
Fe(CN)₆³⁻
>>> print(ferricyanide.latex_name + ", " + ferricyanide.html_name)
Fe(CN)_{6}^{3-}, Fe(CN)<sub>6</sub><sup>3-</sup>
>>> print('%.3f' % ferricyanide.mass)
211.955as you see, in composition, the atomic numbers (and 0 for charge) is used as keys and the count of each kind became respective value.
>>> from chempy import balance_stoichiometry # Main reaction in NASA's booster rockets:
>>> reac, prod = balance_stoichiometry({'NH4ClO4', 'Al'}, {'Al2O3', 'HCl', 'H2O', 'N2'})
>>> from pprint import pprint
>>> pprint(dict(reac))
{'Al': 10, 'NH4ClO4': 6}
>>> pprint(dict(prod))
{'Al2O3': 5, 'H2O': 9, 'HCl': 6, 'N2': 3}
>>> from chempy import mass_fractions
>>> for fractions in map(mass_fractions, [reac, prod]):
... pprint({k: '{0:.3g} wt%'.format(v*100) for k, v in fractions.items()})
...
{'Al': '27.7 wt%', 'NH4ClO4': '72.3 wt%'}
{'Al2O3': '52.3 wt%', 'H2O': '16.6 wt%', 'HCl': '22.4 wt%', 'N2': '8.62 wt%'}ChemPy can also balance reactions where the reacting species are more complex and are better described in other terms than their molecular formula. A silly, yet illustrative example would be how to make pancakes without any partially used packages:
>>> substances = {s.name: s for s in [
... Substance('pancake', composition=dict(eggs=1, spoons_of_flour=2, cups_of_milk=1)),
... Substance('eggs_6pack', composition=dict(eggs=6)),
... Substance('milk_carton', composition=dict(cups_of_milk=4)),
... Substance('flour_bag', composition=dict(spoons_of_flour=60))
... ]}
>>> pprint([dict(_) for _ in balance_stoichiometry({'eggs_6pack', 'milk_carton', 'flour_bag'},
... {'pancake'}, substances=substances)])
[{'eggs_6pack': 10, 'flour_bag': 2, 'milk_carton': 15}, {'pancake': 60}]ChemPy can even handle reactions with linear dependencies (underdetermined systems), e.g.:
>>> pprint([dict(_) for _ in balance_stoichiometry({'C', 'O2'}, {'CO2', 'CO'})]) # doctest: +SKIP
[{'C': x1 + 2, 'O2': x1 + 1}, {'CO': 2, 'CO2': x1}]the x1 object above is an instance of SymPy's Symbol. If we prefer to get a solution
with minimal (non-zero) integer coefficients we can pass underdetermined=None:
>>> pprint([dict(_) for _ in balance_stoichiometry({'C', 'O2'}, {'CO2', 'CO'}, underdetermined=None)])
[{'C': 3, 'O2': 2}, {'CO': 2, 'CO2': 1}]note however that even though this solution is in some sense "canonical",
it is merely one of an inifite number of solutions (x1 from earlier may be any integer).
>>> from chempy import Equilibrium
>>> from sympy import symbols
>>> K1, K2, Kw = symbols('K1 K2 Kw')
>>> e1 = Equilibrium({'MnO4-': 1, 'H+': 8, 'e-': 5}, {'Mn+2': 1, 'H2O': 4}, K1)
>>> e2 = Equilibrium({'O2': 1, 'H2O': 2, 'e-': 4}, {'OH-': 4}, K2)
>>> coeff = Equilibrium.eliminate([e1, e2], 'e-')
>>> coeff
[4, -5]
>>> redox = e1*coeff[0] + e2*coeff[1]
>>> print(redox)
32 H+ + 4 MnO4- + 20 OH- = 26 H2O + 4 Mn+2 + 5 O2; K1**4/K2**5
>>> autoprot = Equilibrium({'H2O': 1}, {'H+': 1, 'OH-': 1}, Kw)
>>> n = redox.cancel(autoprot)
>>> n
20
>>> redox2 = redox + n*autoprot
>>> print(redox2)
12 H+ + 4 MnO4- = 6 H2O + 4 Mn+2 + 5 O2; K1**4*Kw**20/K2**5Functions and objects useful
for working with units are available from the chempy.units module. Here is an
example of how ChemPy can check consistency of units:
>>> from chempy import Reaction
>>> r = Reaction.from_string("H2O -> H+ + OH-; 1e-4/M/s")
Traceback (most recent call last):
...
ValueError: Unable to convert between units of "1/M" and "dimensionless"
>>> r = Reaction.from_string("H2O -> H+ + OH-; 1e-4/s")
>>> from chempy.units import to_unitless, default_units as u
>>> to_unitless(r.param, 1/u.minute)
0.006right now the .units module wraps the quantities package with some minor
additions and work-arounds. However, there is no guarantee that the underlying
package will not change in a future version of ChemPy (there are many packages
for dealing with units in the scientific Python ecosystem).
If we want to predict pH of a bicarbonate solution we simply just need pKa and pKw values:
>>> from collections import defaultdict
>>> from chempy.equilibria import EqSystem
>>> eqsys = EqSystem.from_string("""HCO3- = H+ + CO3-2; 10**-10.3
... H2CO3 = H+ + HCO3-; 10**-6.3
... H2O = H+ + OH-; 10**-14/55.4
... """) # pKa1(H2CO3) = 6.3 (implicitly incl. CO2(aq)), pKa2=10.3 & pKw=14
>>> arr, info, sane = eqsys.root(defaultdict(float, {'H2O': 55.4, 'HCO3-': 1e-2}))
>>> conc = dict(zip(eqsys.substances, arr))
>>> from math import log10
>>> print("pH: %.2f" % -log10(conc['H+']))
pH: 8.30here is another example for ammonia:
>>> from chempy import Equilibrium
>>> from chempy.chemistry import Species
>>> water_autop = Equilibrium({'H2O'}, {'H+', 'OH-'}, 10**-14) # unit "molar" assumed
>>> ammonia_prot = Equilibrium({'NH4+'}, {'NH3', 'H+'}, 10**-9.24) # same here
>>> substances = [Species.from_formula(f) for f in 'H2O OH- H+ NH3 NH4+'.split()]
>>> eqsys = EqSystem([water_autop, ammonia_prot], substances)
>>> print('\n'.join(map(str, eqsys.rxns))) # "rxns" short for "reactions"
H2O = H+ + OH-; 1e-14
NH4+ = H+ + NH3; 5.75e-10
>>> init_conc = defaultdict(float, {'H2O': 1, 'NH3': 0.1})
>>> x, sol, sane = eqsys.root(init_conc)
>>> assert sol['success'] and sane
>>> print(', '.join('%.2g' % v for v in x))
1, 0.0013, 7.6e-12, 0.099, 0.0013ChemPy collects equations and utility functions for working with concepts such as ionic strength:
>>> from chempy.electrolytes import ionic_strength
>>> ionic_strength({'Fe+3': 0.050, 'ClO4-': 0.150}) == .3
Truenote how ChemPy parsed the charges from the names of the substances. There are also e.g. empirical equations and convenience classes for them available, e.g.:
>>> from chempy.henry import Henry
>>> kH_O2 = Henry(1.2e-3, 1800, ref='carpenter_1966')
>>> print('%.1e' % kH_O2(298.15))
1.2e-03to get more information about e.g. this class, you may can look at the API documentation.
A common task when modelling problems in chemistry is to investigate the time dependence of a system. This branch of study is known as chemical kinetics, and ChemPy has some classes and functions for working with such problems:
>>> from chempy import ReactionSystem # The rate constants below are arbitrary
>>> rsys = ReactionSystem.from_string("""2 Fe+2 + H2O2 -> 2 Fe+3 + 2 OH-; 42
... 2 Fe+3 + H2O2 -> 2 Fe+2 + O2 + 2 H+; 17
... H+ + OH- -> H2O; 1e10
... H2O -> H+ + OH-; 1e-4""") # "[H2O]" = 1.0 (actually 55.4 at RT)
>>> from chempy.kinetics.ode import get_odesys
>>> odesys, extra = get_odesys(rsys)
>>> from collections import defaultdict
>>> import numpy as np
>>> tout = sorted(np.concatenate((np.linspace(0, 23), np.logspace(-8, 1))))
>>> c0 = defaultdict(float, {'Fe+2': 0.05, 'H2O2': 0.1, 'H2O': 1.0, 'H+': 1e-2, 'OH-': 1e-12})
>>> result = odesys.integrate(tout, c0, atol=1e-12, rtol=1e-14)
>>> import matplotlib.pyplot as plt
>>> fig, axes = plt.subplots(1, 2, figsize=(12, 5))
>>> for ax in axes:
... _ = result.plot(names=[k for k in rsys.substances if k != 'H2O'], ax=ax)
... _ = ax.legend(loc='best', prop={'size': 9})
... _ = ax.set_xlabel('Time')
... _ = ax.set_ylabel('Concentration')
>>> _ = axes[1].set_ylim([1e-13, 1e-1])
>>> _ = axes[1].set_xscale('log')
>>> _ = axes[1].set_yscale('log')
>>> _ = fig.tight_layout()
>>> _ = fig.savefig('examples/kinetics.png', dpi=72)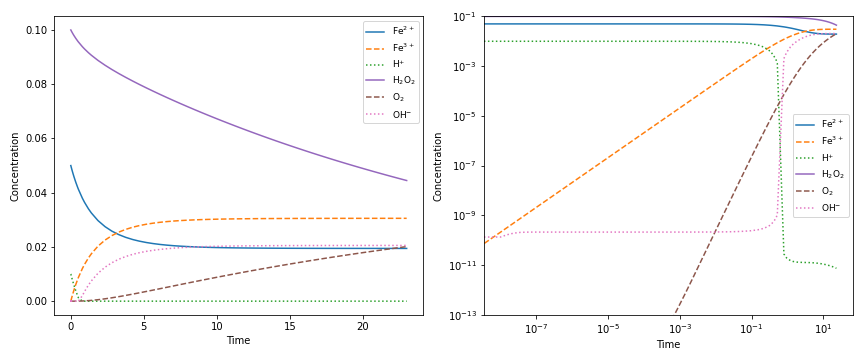
One of the fundamental tasks in science is the careful collection of data about the world around us. ChemPy contains a growing collection of parametrizations from the scientific literature with relevance in chemistry. Here is how you use one of these formulations:
>>> from chempy import Substance
>>> from chempy.properties.water_density_tanaka_2001 import water_density as rho
>>> from chempy.units import to_unitless, default_units as u
>>> water = Substance.from_formula('H2O')
>>> for T_C in (15, 25, 35):
... concentration_H2O = rho(T=(273.15 + T_C)*u.kelvin, units=u)/water.molar_mass(units=u)
... print('[H2O] = %.2f M (at %d °C)' % (to_unitless(concentration_H2O, u.molar), T_C))
...
[H2O] = 55.46 M (at 15 °C)
[H2O] = 55.35 M (at 25 °C)
[H2O] = 55.18 M (at 35 °C)Using only a web-browser (and an internet connection) it is possible to explore the notebooks here: (by the courtesy of the people behind mybinder)
If you make use of ChemPy in e.g. academic work you may cite the following peer-reviewed publication:
Depending on what underlying solver you are using you should also cite the appropriate paper (you can look at the list of references in the JOSS article). If you need to reference, in addition to the paper, a specific point version of ChemPy (for e.g. reproducibility) you can get per-version DOIs from the zendodo archive:
The source code is Open Source and is released under the very permissive "simplified (2-clause) BSD license". See LICENSE for further details.
Contributors are welcome to suggest improvements at https://github.com/bjodah/chempy (see further details here).
- Björn I. Dahlgren, contact:
- gmail address: bjodah