{kind=link}
In case you are interested, you can open the project report.
The reason behind this project is to create my own parallelized enumeration algorithm for all ungapped nucleotide sequence motifs in a set of sequences.
While the testgen.py test generator has been also parallelized, the msa.py multiple sequence alignment, which uses ClustalOmega, has not been parallelized, as it is not in the scope of this project.
The approach we used is load balancing of threads with shared memory. It consists of multiple steps, which are:
- Create the profile matrix by dividing its columns among the available threads, with each thread going over all the sequences at the assigned indices for the thread;
- Anchoring the probabilities that are below a certain threshold, in order to create a probability vector which contains at each index the character(s) that has a probability greater than the threshold;
- Identify the ranges of indices that represent ungapped sub-sequences, which are at least of the required motif length or greater;
- Distribute the threads among subsets of each range of size similar to the length of the requested motif. That way we can get every possible motif starting from each possible index in that range and continuing until its length is as the length required. Each motif will have a scoring of 1, when empty, and it will be multiplied by
p(char)/(1/len(alphabet)); - Calculate the distance between each pair of motifs by taking the absolute value of their difference,
abs(score(motif1)-score(motif2)). Then keep only the motifs that are approximately distant apart by a metric d with a percentage error of e.
python3 testgen.py 10 1000000 1000 > fasta/test_2.py| argument | example | name |
|---|---|---|
| 1 | 10 | number of processors |
| 2 | 1000000 | nucleotide sequence length |
| 3 | 1000 | number of nucleotide sequences |
python3 msa.py .out fasta/test_2.py| argument | example | name |
|---|---|---|
| 1 | .out | output file |
| 2+ | fasta/test_2.py | input files |
Please note, this is serial. It is done by running ClustalOmega on the fastas. If your sequences are of the same length and you prefer not wasting your time running this, then don't.
python3 modis.py .out 64 10 20 0.3 100 0.05| argument | example | name |
|---|---|---|
| 1 | .out | input file |
| 2 | 64 | number of processors |
| 3 | 10 | motif length |
| 4 | 20 | max number of motifs in result |
| 5 | 0.3 | nucleotide probability threshold |
| 6 | 100 | distance between motifs |
| 7 | 0.05 | percentage of accepted error in distance |
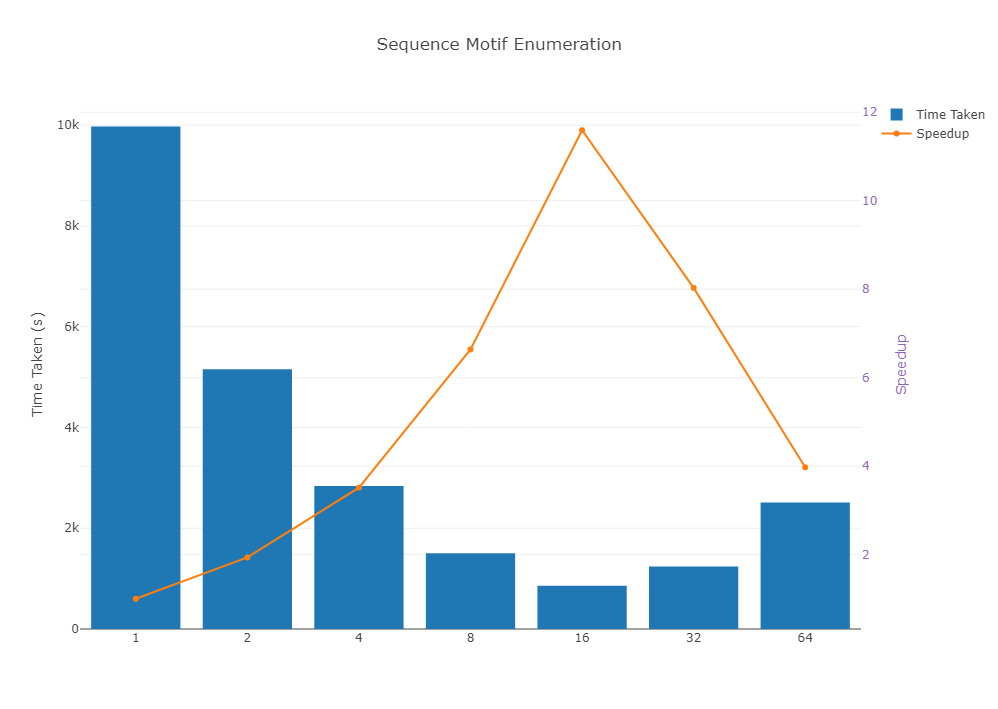
After running the code on a dataset of size 1,000 sequences of size 1,000,000, the following speedup was achieved.