Run on a local system
Create a folder and download there the previous compressed FAST5 dataset, then uncompress this file:
$ mkdir tutorial
$ cd tutorial
$ tar zxvf test_fast5.tar.gz
Copy the HPG Pore script, the jar and dynamic library in the folder tutorial (where you have downloaded and uncompressed the dataset). So, you should have these files in that folder:
test_fast5
test_fast5.tar.gz
hpg-pore-0.1.0-jar-with-dependencies.jar
libhpgpore.so
hpg-pore.sh
To run the command stats, first you should create a folder where to save the output results:
$ mkdir out-stats
$ ./hpg-pore.sh stats --in test_fast5 --out out-stats
The command stats creates a file summary.txt containing several statistics and a folder per run to save histograms and graphs. In our example:
$ ls -ltr out-stats/
total 8
drwxrwxr-x 2 jtarraga jtarraga 4096 May 11 12:08 d5c085dc93da5740a906ccfd86aad93c2f0a44c8
-rw-rw-r-- 1 jtarraga jtarraga 1061 May 11 12:19 summary.txt
The content of the file summary.txt:
$ cat out-stats/summary.txt
-----------------------------------------------------------------------
Statistics for run d5c085dc93da5740a906ccfd86aad93c2f0a44c8
-----------------------------------------------------------------------
Template:
Num. seqs: 69
Num. nucleotides: 341458
Mean read length: 4948
Min. read length: 42
Max. read length: 17420
Nucleotides content:
A: 80450 (23.56 %)
T: 86374 (25.30 %)
G: 90780 (26.59 %)
C: 83854 (24.56 %)
N: 0 (0.00 %)
GC: 51.14 %
Mean read quality: 37
Complement:
Num. seqs: 26
Num. nucleotides: 144914
Mean read length: 5573
Min. read length: 830
Max. read length: 9544
Nucleotides content:
A: 35648 (24.60 %)
T: 36154 (24.95 %)
G: 37993 (26.22 %)
C: 35119 (24.23 %)
N: 0 (0.00 %)
GC: 50.45 %
Mean read quality: 37
2D:
Num. seqs: 20
Num. nucleotides: 136257
Mean read length: 6812
Min. read length: 1916
Max. read length: 10090
Nucleotides content:
A: 34325 (25.19 %)
T: 34088 (25.02 %)
G: 34143 (25.06 %)
C: 33701 (24.73 %)
N: 0 (0.00 %)
GC: 49.79 %
Mean read quality: 42
And the histograms and images generated by the the run d5c085dc93da5740a906ccfd86aad93c2f0a44c8:
$ ls -ltr out-stats/d5c085dc93da5740a906ccfd86aad93c2f0a44c8/
total 2216
-rw-rw-r-- 1 jtarraga jtarraga 122091 May 11 13:16 reads_per_channel.jpg
-rw-rw-r-- 1 jtarraga jtarraga 114092 May 11 13:16 yield_per_channel.jpg
-rw-rw-r-- 1 jtarraga jtarraga 143430 May 11 13:16 Template_length_histogram.jpg
-rw-rw-r-- 1 jtarraga jtarraga 136725 May 11 13:16 Complement_length_histogram.jpg
-rw-rw-r-- 1 jtarraga jtarraga 130031 May 11 13:16 2D_length_histogram.jpg
-rw-rw-r-- 1 jtarraga jtarraga 72637 May 11 13:16 Template_quality_histogram.jpg
-rw-rw-r-- 1 jtarraga jtarraga 79887 May 11 13:16 Complement_quality_histogram.jpg
-rw-rw-r-- 1 jtarraga jtarraga 72701 May 11 13:16 2D_quality_histogram.jpg
-rw-rw-r-- 1 jtarraga jtarraga 99084 May 11 13:16 Template_yield.jpg
-rw-rw-r-- 1 jtarraga jtarraga 100973 May 11 13:16 Complement_yield.jpg
-rw-rw-r-- 1 jtarraga jtarraga 95601 May 11 13:16 2D_yield.jpg
-rw-rw-r-- 1 jtarraga jtarraga 101578 May 11 13:16 Template_quality_per_pos.jpg
-rw-rw-r-- 1 jtarraga jtarraga 110502 May 11 13:16 Complement_quality_per_pos.jpg
-rw-rw-r-- 1 jtarraga jtarraga 103784 May 11 13:16 2D_quality_per_pos.jpg
-rw-rw-r-- 1 jtarraga jtarraga 118299 May 11 13:16 Template_content_per_pos.jpg
-rw-rw-r-- 1 jtarraga jtarraga 135003 May 11 13:16 Complement_content_per_pos.jpg
-rw-rw-r-- 1 jtarraga jtarraga 133907 May 11 13:16 2D_content_per_pos.jpg
-rw-rw-r-- 1 jtarraga jtarraga 90795 May 11 13:16 Template_GC_histogram.jpg
-rw-rw-r-- 1 jtarraga jtarraga 91373 May 11 13:16 Complement_GC_histogram.jpg
-rw-rw-r-- 1 jtarraga jtarraga 101286 May 11 13:16 2D_GC_histogram.jpg
You can extract the sequences in format FastQ and FASTA by executing the commands fastq and fasta respectively, e.g.: extracting sequences in FastQ format:
$ mkdir out-fastq
$ ./hpg-pore.sh fastq --in test_fast5 --out out-fastq
A folder is created per run, in our case, we have one run: d5c085dc93da5740a906ccfd86aad93c2f0a44c8
$ ls -ltr out-fastq/d5c085dc93da5740a906ccfd86aad93c2f0a44c8/
total 1236
-rw-rw-r-- 1 jtarraga jtarraga 684625 May 11 14:28 template.fq
-rw-rw-r-- 1 jtarraga jtarraga 290472 May 11 14:28 complement.fq
-rw-rw-r-- 1 jtarraga jtarraga 273008 May 11 14:28 2D.fq
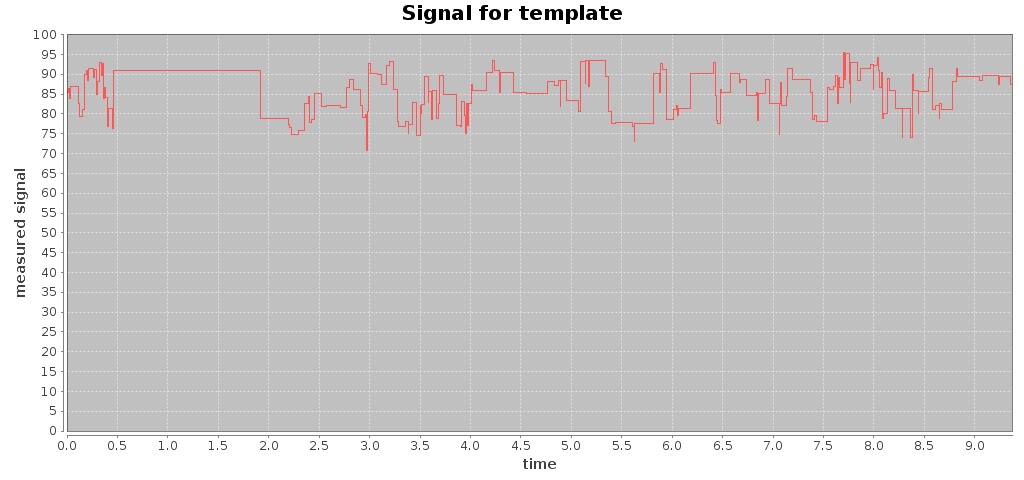
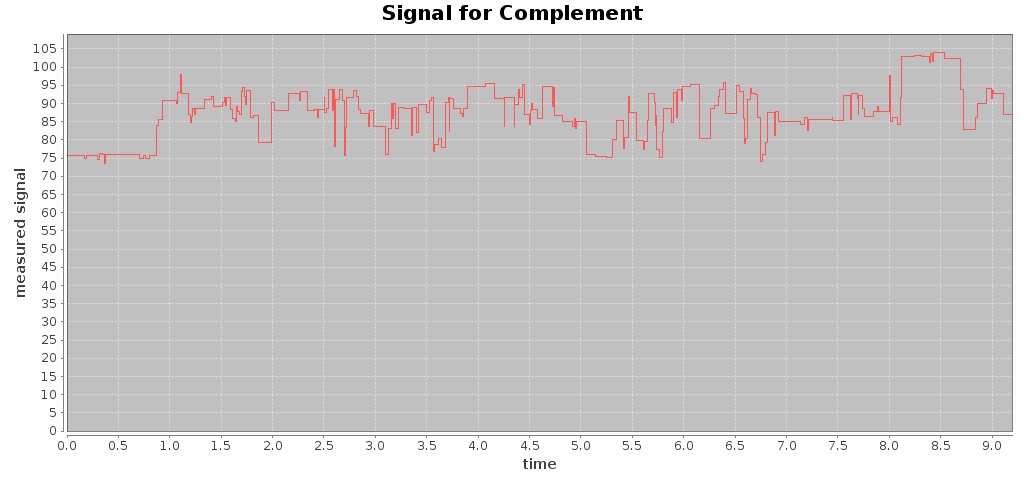
For a given Fast5 file you can also extract raw data of the electronic signal measured (by executing the command events) and plot the signal over time (by using the command signal), e.g. plotting the signal for the first 10 seconds:
$ mkdir out-signal
$ /hpg-pore.sh signal --in test_fast5/LomanLabz_PC_E.coli_MG1655_ONI_3058_1_ch15_file33_strand.fast5 --out out-signal --min 0 --max 10
For this Fast5 file, two signals are plotted for the template and complement sequences:
$ ls -ltr out-signal/
total 244
-rw-rw-r-- 1 jtarraga jtarraga 116307 May 11 14:14 template_signal.jpg
-rw-rw-r-- 1 jtarraga jtarraga 121762 May 11 14:14 complement_signal.jpg