A new AI model trained and tested with fresh updated dataset of small Non-coding RNA (ncRNA or sncRNA) sequences to resolve efficiently the classification of small non-coding RNA. Biological experimental methods for identifying ncRNA families are not only time-consuming and labor-intensive but also expensive, making them impractical for the demands of high-throughput technology.
| Method/Model | Accuracy | Sensitivity | Precision | F-score | MCC |
|---|---|---|---|---|---|
| RNAcon | 0.3737 | 0.3787 | 0.4500 | 0.3605 | 0.3341 |
| GeaPPLE | 0.6487 | 0.6684 | 0.7325 | 0.7050 | 0.6857 |
| nRC | 0.6960 | 0.6889 | 0.6878 | 0.6878 | 0.6627 |
| ncRFP | 0.7972 | 0.7878 | 0.7904 | 0.7883 | 0.7714 |
| ncDLRES | 0.8430 | 0.8344 | 0.8419 | 0.8407 | 0.8335 |
| ncDENSE | 0.8687 | 0.8677 | 0.8703 | 0.8667 | 0.8574 |
| --> NCC | 0.9897 | 0.9870 | 0.9892 | 0.9880 | 0.9889 |
| MncR | > 97% | - | - | - | - |
| Functions | Files |
|---|---|
| Data collection functions | rfam_query.py |
| Data Analysis | Analysis.ipynb |
| Data transformation | ncc_DataTransform.py |
| AI Models | ncc_Model.py |
| Training and testing the model | ncc_TrainTest.py |
To collect datasets from Rfam database and assemble the main used dataset you will find methods in rfam_query.py file
# Update if you need more or less RNA families to be downloaded form Rfam db
def get_RNA_Families_in_interest() -> []:
return [
'Cis-reg; IRES;',
'Cis-reg; leader;',
'Cis-reg; riboswitch;',
'Cis-reg; riboswitch;',
'Gene; ribozyme;',
'Gene; rRNA;',
'Gene; miRNA;',
'Gene; snRNA; snoRNA; CD-box;',
'Gene; snRNA; snoRNA; HACA-box;',
'Gene; snRNA; snoRNA; scaRNA;',
'Gene; tRNA;',
'Intron;'
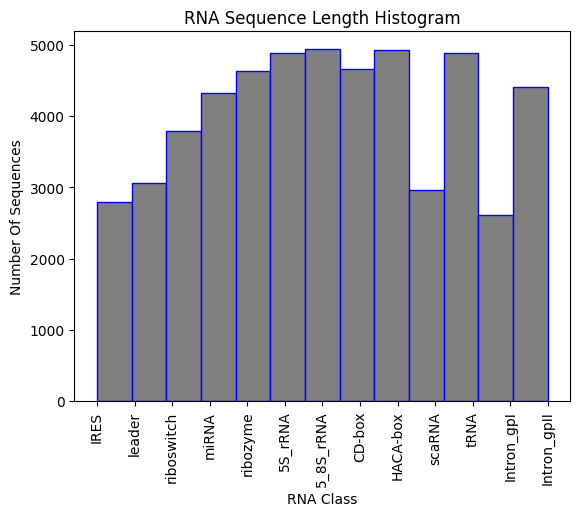
]If a Jupiter Notebook with some statictic analysis of the dataset that can help finalize the data input of the AI model. The final dataset has more than 50.000 labeld RNA sequences in fasta format as shown bellow:
>IRES
ATACCTTTCTCGGCCTTTTGGCTAAGATCAAGTGTAGTATCTGTTCTTATCAGTTTAATATCTGATACGTGGGCCA ...
>tRNA
GCACCACTCTGGCCTTTTGGCTTAGATCAAGTGTAGTATCTGTTCTTATTAGTTTAACCACTAATATGGTCGCACC ...
>tRNA
ATACCTTTCTCGGCCTTTTGGCTAAGATCAAGTGTAGTATCTGTTTTTATCAGTTTAATATCTGATATGTGGTCCA ...
>riboswitch
ATTACTTCTCAGCCTTTTGGCTAAGATCAAGTGTAATAAATCTCATTGTGCTTTATGCCTAATGTGTGCTTATATT ...
>HACA-box
CCAGCTCTCTTTGCCTTTTGGCTTAGATCAAGTGTAGTATCTGTTCTTTTCAGTTTAATCTCTGAAAGTGTTCTAA ...
>tRNA
ACAGCTGATGCCGCAGCTACACTATGTATTAATCGGATTTTTGAACTTGGAGTACGGTTCTGGAGCTTGCTCCACC ...
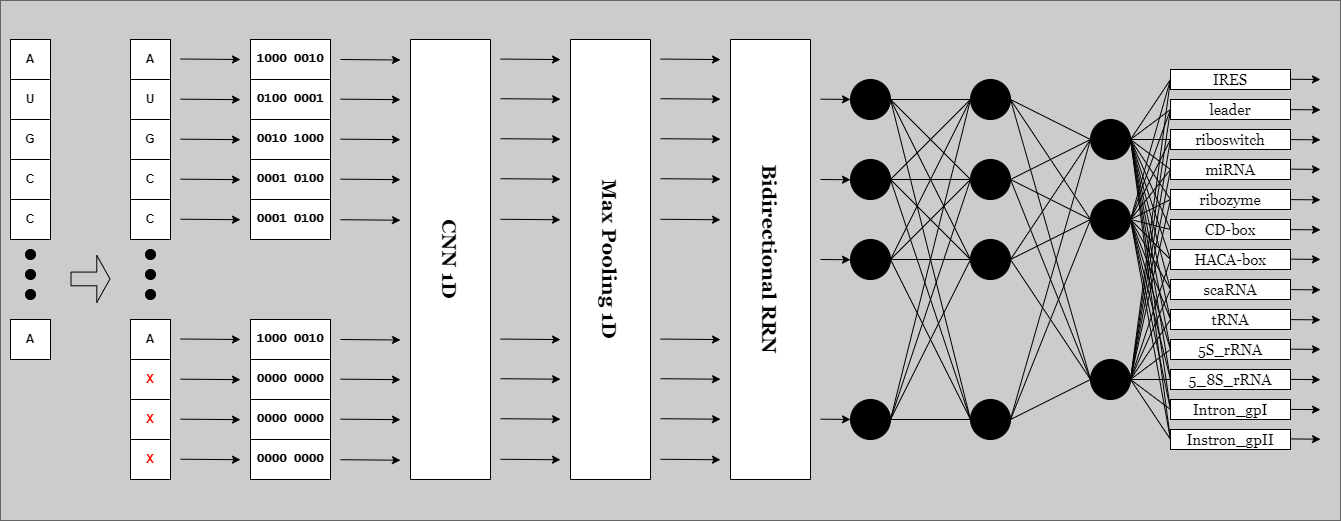
Padding, cutting and encoding the RNA sequences before loading them to AI model. If you and to change the encoding method edit this file. One-hot encoding is used.
# Ribisome encoding
# --------------------------------------
A_rep_8d = [1, 0, 0, 0, 0, 0, 1, 0]
U_rep_8d = [0, 1, 0, 0, 0, 0, 0, 1]
G_rep_8d = [0, 0, 1, 0, 1, 0, 0, 0]
C_rep_8d = [0, 0, 0, 1, 0, 1, 0, 0]
X_rep_8d = [0, 0, 0, 0, 0, 0, 0, 0]The keras model used for this task. Consists of an Biderectional RRN in the input and Densenet CNN.
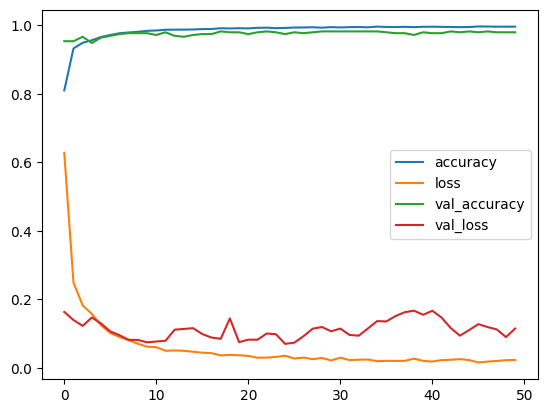
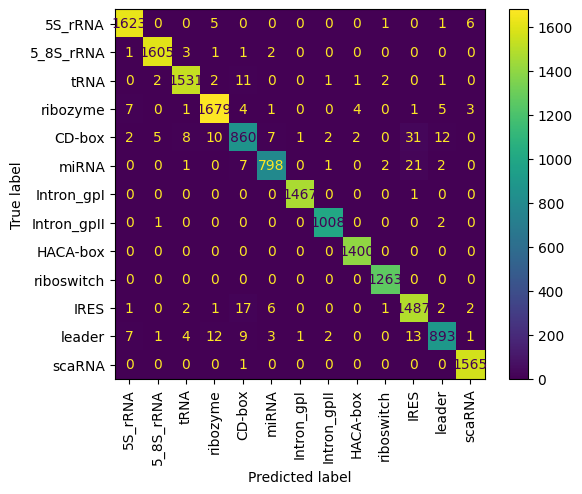
A jupiter Notepad for training evaluating/tasting the selected model and some metrics along.
 |
 |
- python
- docker - Docker SDK for Python
- wget
- fastaparser - A Python FASTA file Parser and Writer
NEED TO UPDATE
Rfam database is a collection of RNA families, each represented by multiple sequence alignments, consensus secondary structures and covariance models