






Reads to Genes, or r2g, is a computationally lightweight and homology-based pipeline that allows rapid identification of genes or gene families from raw sequence databases in the absence of an assembly, by taking advantage of over 44.3 petabases of sequencing data for all kinds of species deposited in Sequence Read Archive hosted by National Center for Biotechnology Information, which can be effectively run on most common computers without high-end specs.
The GUI wrapper r2g GUI now is released. Please visit here if you prefer a graphic user interface (GUI) for r2g. The following methods are for installing command line interface (CLI) for r2g. Please note that GUI is still under developing, and CLI is more stable than GUI.
Please follow the instruction here to download and install Docker based on your operating system before running the Docker image. For Windows users, please check here to configure the Docker if it is your first time to use it.
This installation method is recommended as it is compatible with most common operating systems including Linux, macOS and Windows.
Then, pull the r2g Docker image with all required software packages installed and configured by one command as follows:
docker pull yangwu91/r2g:latest
Now, you are good to go.
For Linux users, r2g can be installed by Conda as follows. Of course miniconda3 (recommended) or anaconda needs to be installed first.
# Install miniconda3:
wget https://repo.anaconda.com/miniconda/Miniconda3-latest-Linux-x86_64.sh
sh Miniconda3-latest-Linux-x86_64.sh
# Set up bioconda channel (or its mirrors):
conda config --add channels defaults
conda config --add channels bioconda
conda config --add channels conda-forge
# Install r2g:
conda install -c yangwu91 r2gAfter that, Google Chrome web browser and the corresponding version of ChromeDriver (or selenium/standalone-chrome Docker image) need to be installed.
In the future, I plan to create a pull request to the Bioconda recipes.
Progress:
- Build Homebrew Formula
- Init a pull request to the
brewsci/bioTap. - Be permitted by the
brewsci/bioTap.
Since the r2g formula is still waiting for the approval from the the brewsci/bio Tap, macOS users can download the r2g formula and add it manually on your local computer.
# Install Homebrew and add the tap
/bin/bash -c "$(curl -fsSL https://raw.githubusercontent.com/Homebrew/install/master/install.sh)"
brew tap brewsci/bio
# Download the r2g formula and put it in the correct directory:
/usr/local/Cellar/curl/7.72.0/bin/curl -o /usr/local/Homebrew/Library/Taps/brewsci/homebrew-bio/Formula/r2g.rb -fsSL https://raw.githubusercontent.com/yangwu91/r2g/master/brewsci-Formula/r2g.rb
# Install r2g:
brew install r2g
And then Google Chrome web browser and the corresponding version of ChromeDriver (or selenium/standalone-chrome Docker image) need to be installed.
The r2g required 3 third-party software packages including NCBI SRA Toolkit, Trinity, and Google Chrome web browser with ChromeDriver (or selenium/standalone-chrome Docker image).
-
NCBI SRA Toolkit
-
For Linux and macOS users, it also can be installed using Conda via the Bioconda channel:
conda install -c bioconda sra-tools
If the installed version of SRA Toolkit is above 2.10.3, before the first run you have to execute the follow command:
vdb-config --interactive
Then press
xto set up the default configs. This is a known annoying issue that can't be avoided.
-
-
Trinity
-
Follow the instruction to compile the source code. Please note that Trinity has its own dependencies, including samtools, Python 3 with NumPy, bowtie2, jellyfish, salmon, and trimmomatic. If you are a macOS user while compiling Trinity, please use
gcccompiler instead of nativeclangcomplier on macOS to avoid raising errors. -
For macOS users, Trinity can be installed using Homebrew as well:
brew tap brewsci/bio brew install trinity
-
For Linux users, Trinity can be installed easily using Conda, and you would never worry about other dependencies:
conda install -c bioconda trinity=2.8.5 numpy samtools=1.10
The compatibility of Trinity Version 2.8.5 with r2g has been fully tested, and theoretically, the later versions should work too.
-
-
Google Chrome web browser with ChromeDriver
-
Install Google Chrome web browser and then download the corresponding version of ChromeDriver.
-
Or, you can simply run selenium/standalone-chrome Docker image in background (make sure you have the permission to bind the 4444 port on local host):
docker run -d -p 4444:4444 -v /dev/shm:/dev/shm selenium/standalone-chrome
-
The r2g package has been deposited to PyPI, so it can be installed as follows:
pip install r2g
If these required third-party applications above are installed using Conda, you don't need to take care of it.
If these packages are compiled or downloaded by yourself, either include them in $PATH separately by a command as follows:
# Linux and macOS:
export PATH="$PATH:/path/to/fastq-dump:/path/to/Trinity:/path/to/chromedriver"
# Windows:
set PATH=%PATH%;DRIVER:\path\to\fastq-dump;DRIVER:\path\to\Trinity;DRIVER:\path\to\chromedriver
or follow the prompts to set up the path to the executable files manually before the first run. And then, you are good to go.
The recommended minimal hardware specifications are 2-core CPU and 4 Gb memory, which are satisfied for most common personal computers nowadays.
Detailed usage will be printed by the command:
docker run -it yangwu91/r2g:latest --helpOr:
r2g --helpOptional arguments:
-h, --help show this help message and exit
-V, --version Print the version.
-v, --verbose Print detailed log.
-r [INT], --retry [INT]
Number of times to retry.Enabling it without any numbers will force it to keep retrying. Default: 5.
--cleanup Clean up all intermediate files and only retain the final assembled contig file in FASTA format.
-o DIR, --outdir DIR Specify an output directory. Default: current working directory.
-W DIR, --browser DIR
Temporarily overwrite the local path or the remote address of the chrome webdriver. E.g., /path/to/chromedriver or http://127.0.0.1:4444/wd/hub
-P SCHEME://IP:PORT, --proxy SCHEME://IP:PORT
Set up proxies. Http and socks are allowed, but authentication is not supported yet (still testing).
NCBI options:
-s SRA, --sra SRA Choose SRA accessions (comma-separated without blank space). E.g., "SRX885418" (an SRA experiment) or "SRR1812886,SRR1812887" (SRA runs)
-q SEQUENCE, --query SEQUENCE
Submit either a FASTA file or nucleotide sequences.
-p BLAST, --program BLAST
Specify a BLAST program: tblastn, tblastx, or blastn (including megablast, blastn, and discomegablast). Default: blastn.
-m INT, --max_num_seq INT
Maximum number of aligned sequences to retrieve (the actual number of alignments may be greater than this). Default: 1000.
-e FLOAT, --evalue FLOAT
Expected number of chance matches in a random model. Default: 1e-3.
-c FRAGMENT,OVERLAP, --cut FRAGMENT,OVERLAP
Cut sequences and query them respectively to prevent weaker matches from being ignored. Default: 70,20 (nucleotides), or 24,7 (amino acids)
Trinity options:
-t INT, --CPU INT Number of CPU threads to use. Default: the total number of your computer.
--max_memory RAM Suggest max Gb of memory to use by Trinity. Default: 4G.
--min_contig_length INT
Minimum assembled contig length to report. Default: 150.
--trim [TRIM_PARAM] Run Trimmomatic to qualify and trim reads. Using this option without any parameters will trigger preset settings in Trinity for Trimmomatic. See Trinity for more help. Default: disabled.
--stage {no_trinity,jellyfish,inchworm,chrysalis,butterfly}
Stop Trinity after the stage you chose. Default: butterfly (the final stage).
While executing the Docker image, some specific options are required: -v /dev/shm:/dev/shm, -v /path/to/your/workspace:/workspace, and -u $UID.
- The option
-v /dev/shm:/dev/shmshares host's memory to avoid applications crashing inside a Docker container.
-
The option
-v /path/to/your/workspace:/workspacemounts the local directory/path/to/your/workspace(specify your own) to the working directory/workspace(don't change it) inside a Docker container, which is the input and output directory. -
The option
-u $UIDsets the owner of the Docker outputs. Ignoring it will raise permission errors.
Let's say there is a query file in FASTA format named QUERY.fasta in the folder /home/user/r2g_workspace/. As a result, the the simplest full command to run a Docker image should be:
docker run -it -v /dev/shm:/dev/shm -v /home/user/r2g_workspace:/workspace -u $UID yangwu91/r2g:latest -o OUTPUT -q QUERY.fasta -s SRXNNNNNNAfter that, you can check out the results in the folder /home/user/r2g_workspace/OUTPUT/.
We applied the r2g pipeline to search the gene S6K (i.e. AAEL018120 from Aedes aegypti, which is a well-studied species) in Aedes albopictus SRA experiment SRX885420 (https://www.ncbi.nlm.nih.gov/sra/SRX885420) using the engine blastn. Detailed workflow is described as follows.
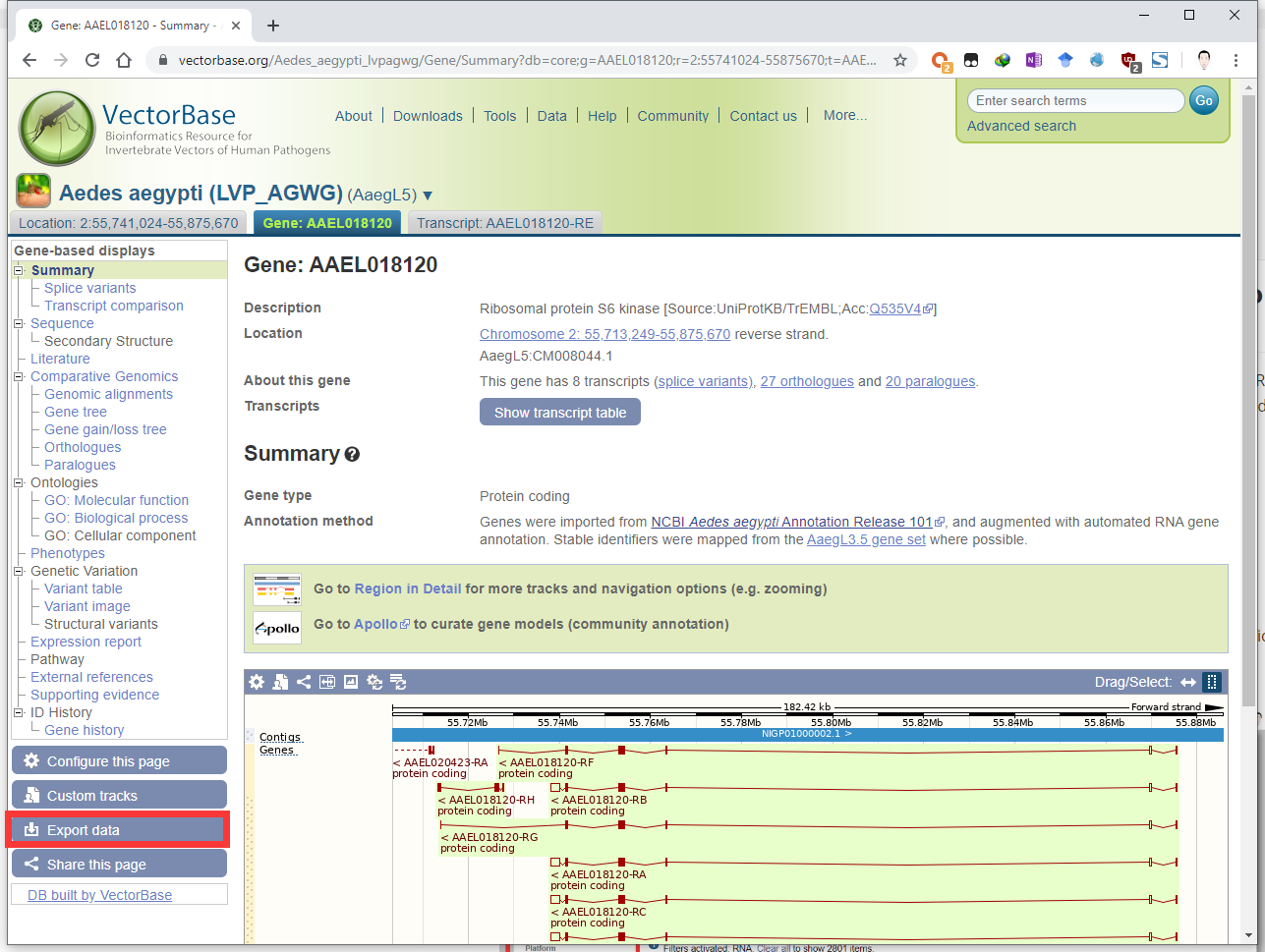
Download nucleotide/protein sequences of Aedes aegypti S6K from VectorBase, Ensembl, NCBI or other online databases, and let’s say it was saved as the file /home/user/r2g_orkspace/AAEL018120-RE.S6K.fasta. Aedes aegypti is a well-studied mosquito species.
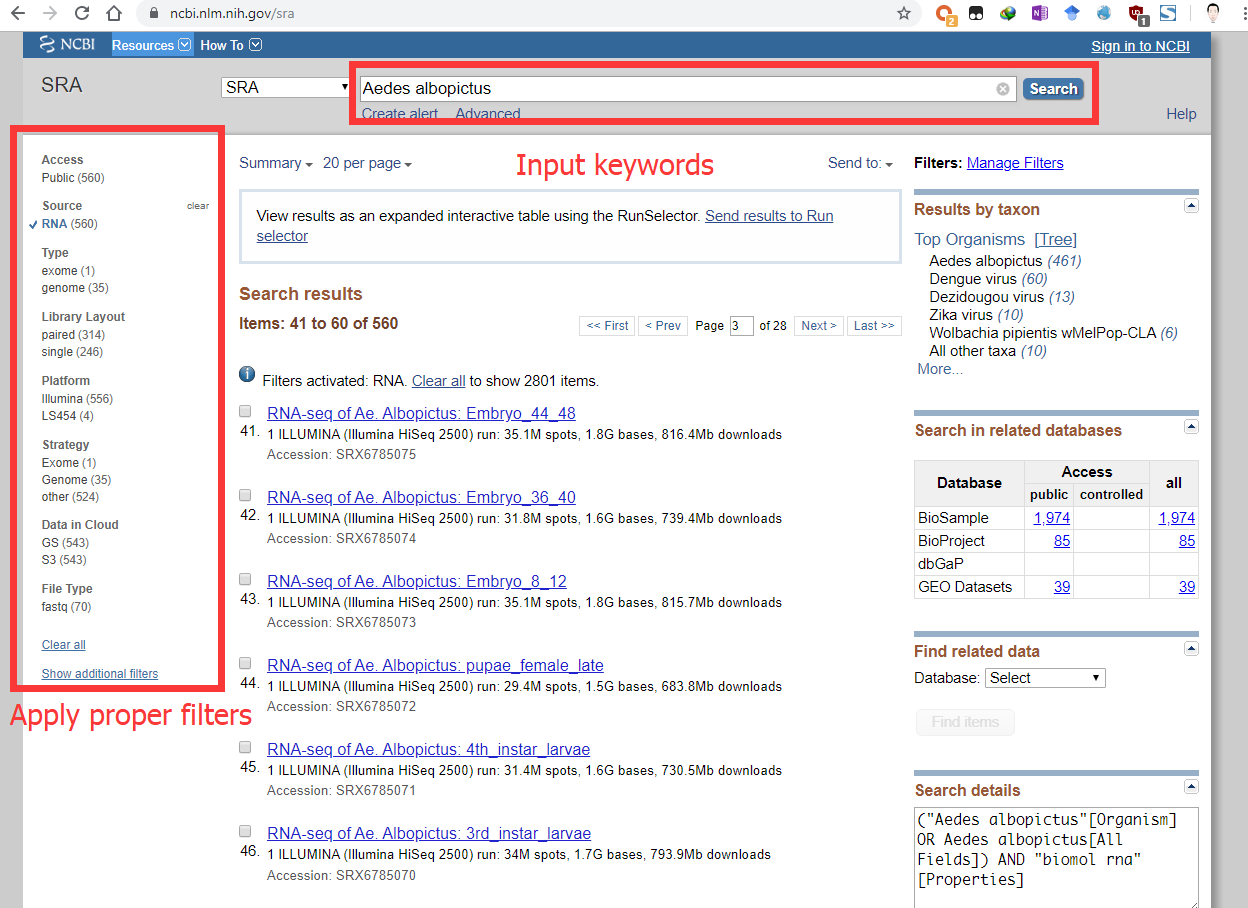
Select a proper SRA experiment for Aedes albopictus (e.g. SRX885420). Some genes only express in specific tissues or at specific time. Make sure the gene you are interested in indeed expresses in the SRA experiment(s) you selected.
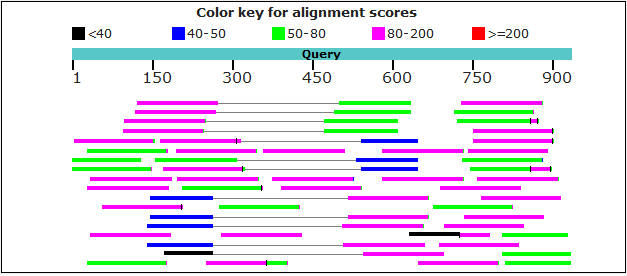
Run the r2g pipeline. Here, we chopped the query (/home/user/r2g_workspace/AAEL018120-RE.S6K.fasta) into 80-base fragments overlapping 50 bases. The command line is as follows:
docker run -it -v /dev/shm:/dev/shm -v /home/user/r2g_workspace:/workspace -u $UID yangwu91/r2g:latest -o /workspace/S6K_q-aae_s-SRX885420_c-80.50_p-blastn -s SRX885420 -q /workspace/AAEL018120-RE.S6K.fasta --cut 80,50 -p blastnOr,
r2g -o /home/user/r2g_workspace/S6K_q-aae_s-SRX885420_c-80.50_p-blastn -s SRX885420 -q /home/user/r2g_workspace/AAEL018120-RE.S6K.fasta --cut 80,50 -p blastnThe sequence file in FASTA format of the predicted Aedes albopictus S6K is in the folder /home/user/r2g_workspace/S6K_q-aae_s-SRX885420_c-80.50_p-blastn/. Please verify the sequences by the PCR amplification or other methods if necessary.