Live Application: https://celldreamer.netlify.app
HuggingFace Space: https://huggingface.co/spaces/RobroKools/CellDreamer-API/tree/main
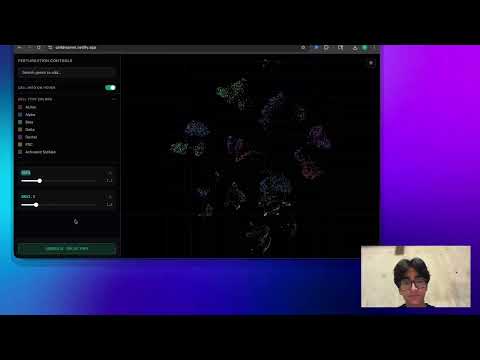
The CellDreamer frontend is a React-based web application that provides an interactive interface for exploring stem cell differentiation trajectories. Users can visualize predicted cell trajectories in a UMAP embedding space, adjust gene expression values to simulate perturbations, and observe how cells move through different cell type clusters (alpha, beta, gamma, etc.) over time. The frontend displays animated trajectories showing how cells differentiate over n=10 steps, providing an intuitive way to explore the model's predictions.
Live Demo: https://celldreamer.netlify.app
We use HuggingFace Spaces to run the world model and handle basic routing logic. The main celldreamer package is located at backend/hf_deployment/celldreamer.
HuggingFace Space: https://huggingface.co/spaces/RobroKools/CellDreamer-API/tree/main
We generate model and cell artifacts to deliver to the frontend and create a powerful animation displaying predicted ductal cell trajectories over n=10 steps. The frontend displays the cell type clusters (alpha, beta, gamma, etc.) that the cell moves towards to differentiate into. We use Render to host our Flask API.
We apply a Dreamer (Hafner et al., 2020) world model with a Recurrent State Space Model (RSSM) and
VAE-based encoder/decoder setup to predict stem cell differentiation gene differentials. We leverage the Panc8 dataset (Stuart et al., 2019) in order to generate synthetic cell differentiation "trajectories,"
where a trajectory is a pair of a given cell's gene expression counts at timestep
In order to turn raw panc8 single-cell data into useable "trajectories," we can't use traditional biological methods like single-cell RNA sequencing since it destroys the cell. So, we use the mathematical method from (Yeo et al., 2020) in order to create a graph of cells, follow them through a random walk via Diffusion Pseudotime (DPT) to calculate cell-to-cell pseudotime distances, and apply topology and causal constraints to generate valid trajectory pairs.
We use K-nearest neighbors to create a graph where each node is a cell and edges represent gene expression similarity, as shown in this line of process.py:
sc.pp.pca(adata, n_comps=50) #create 50 principal components
sc.pp.neighbors(adata, n_neighbors=30, n_pcs=20) # choose the top 20 most biological feature-dense componentsWe use DPT to calculate psuedotimes to assign to each cell from the graph root. The line below of process.py assigns the ductal/graph root cells and then runs DPT to assign psuedotimes from distance from root:
# find step 0 stem cell
try:
root_candidates = np.where(adata.obs['celltype'].str.contains('ductal', case=False))[0]
adata.uns['iroot'] = root_candidates[0] if len(root_candidates) > 0 else 0
except:
adata.uns['iroot'] = 0
sc.tl.dpt(adata)After assigning psuedotimes to each graph, we can use topology + causality constraints to yield trajectory pairs. We specifically filter out jumping between principal components that are not connected and moving backwards (want to train world model from time 0 to time 1, forwards).
The lines below of process.py iterates through the graph and append cell pairs as trajectories using a max time diff = 0.1 as a topology constraint from the DPT calculated psuedotimes.
rows, cols = graph.nonzero()
for i, j in zip(rows, cols):
t_i, t_j = times[i], times[j]
# j is after i and use max time diff is 0.1 for ~similar time diffs
if t_j > t_i and (t_j - t_i) < 0.1:
pairs.append([i, j])| Metric | Value |
|---|---|
| Test Samples | 18,253 |
| Avg. Total Loss | 0.93 ± 0.04 |
| Reconstruction Loss (MSE) | 0.72 |
| Dynamics KL Loss | 10.77 |
| Posterior KL | 10.07 |
CellDreamer demonstrates stable performance across a large held-out test set, with low variance in total loss indicating strong generalization across cell states and pseudotime regions. The reconstruction MSE shows that the encoder–decoder effectively captures high-dimensional gene expression despite scRNA-seq noise and sparsity. Meanwhile, the non-trivial dynamics and posterior KL values suggest that the RSSM learns meaningful stochastic latent transitions rather than collapsing to deterministic dynamics, which is critical for modeling uncertainty and branching in cell fate decisions. Together, these results support the feasibility of learning biologically plausible differentiation dynamics from pseudotime-derived trajectories, even in the absence of true longitudinal single-cell time series.
Please cd backend/hf_deployment from project root and run data collection
chmod +x celldreamer/scripts/data.sh
bash celldreamer/scripts/data.shthen training:
chmod +x celldreamer/scripts/train.sh
bash celldreamer/scripts/train.sh celldreamer/config/train_config.ymland evaluation:
chmod +x celldreamer/scripts/evaluate.sh
bash celldreamer/scripts/evaluate.sh celldreamer/config/evaluate_config.ymlHafner, D., Lillicrap, T., Ba, J., & Norouzi, M. (2020). Dream to Control: Learning Behaviors by Latent Imagination. arXiv [Cs.LG]. http://arxiv.org/abs/1912.01603
Stuart, T., et al. (2019). Comprehensive Integration of Single-Cell Data. Cell, 177(7), 1888-1902. doi:10.1016/j.cell.2019.05.031
Yeo, G. H. T., Saksena, S. D., & Gifford, D. K. (2020). [Title of the manuscript]. bioRxiv. https://doi.org/10.1101/2020.08.26.269332