

{kind=link}
CIRCexplorer is a combined strategy to identify junction reads from back spliced exons and intron lariats.
Version: 1.1.10
Last Modified: 2016-7-14
Authors: Xiao-Ou Zhang (zhangxiaoou@picb.ac.cn), Li Yang (liyang@picb.ac.cn)
Maintainer: Xu-Kai Ma (maxukai@picb.ac.cn)
Download the latest stable version of CIRCexplorer
To see what has changed in recent versions of CIRCexplorer, see the CHANGELOG.
After one year's tensive development, we have upgraded and extended CIRCexplorer to a new version -- CIRCexplorer2, with many improvements and lots of new features. Welcome to use and cite it!
NEWS: CIRCpedia is an integrative database, aiming to annotating alternative back-splicing and alternative splicing in circRNAs across different cell lines. Welcome to use!
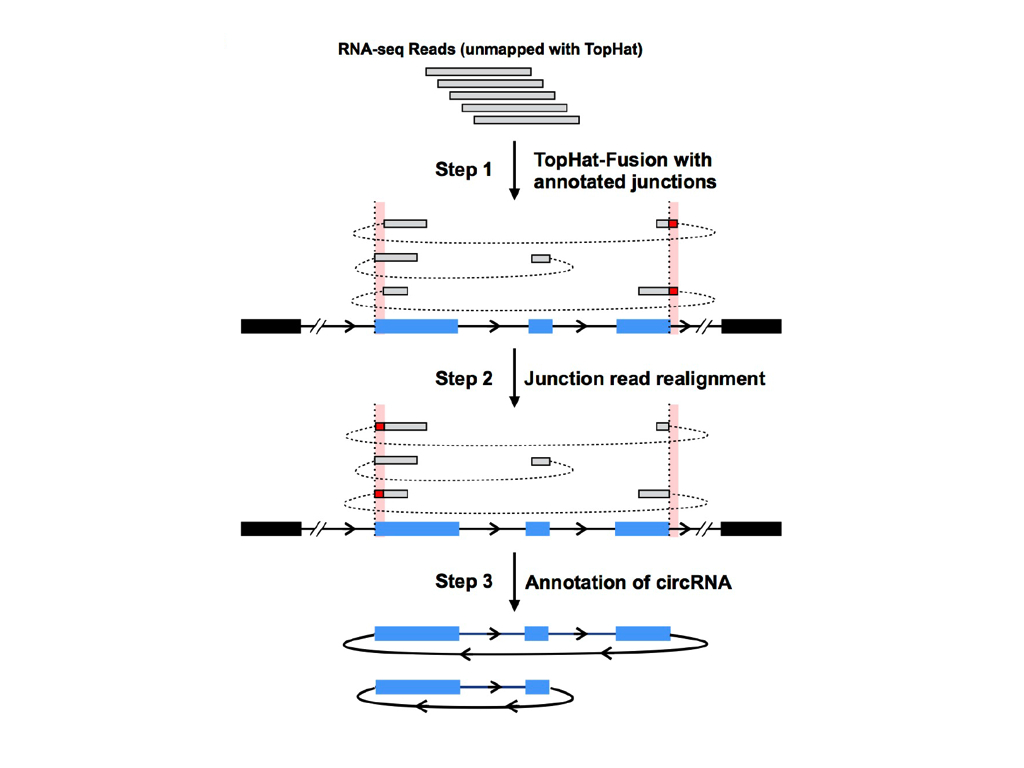
CIRCexplorer is now only a circular RNA annotating tool, and it parses fusion junction information from mapping results of other aligners. The result of circular RNA annotating is directly dependent on the mapping strategy of aligners. Different aligners may have different circular RNA annotations. CIRCexplorer is only in charge of annotating circRNA junctions according to the gene annotation. More functions could be found in CIRCexplorer2.
- TopHat & TopHat-Fusion
- TopHat 2.0.9
- TopHat-Fusion included in TopHat 2.0.9
- STAR (optional)
- STAR 2.4.0j
- bedtools
- pysam >=0.8.2
- docopt
- interval (If you use the old version of CIRCexplorer, you require to install the interval package from https://github.com/kepbod/interval. For the latest version of CIRCexplorer, you won't install it by yourself.)
The poly(A)−/ribo− RNA-seq is recommended. If you want to obtain more circular RNAs, RNase R treatment could be performed.
CIRCexplorer was originally developed as a circular RNA analysis toolkit supporting TopHat & TopHat-Fusion. After version 1.1.0, it also supports STAR.
To obtain junction reads for circular RNAs, two-step mapping strategy was exploited:
- Multiple mapping with TopHat
tophat2 -a 6 --microexon-search -m 2 -p 10 -G knownGene.gtf -o tophat hg19_bowtie2_index RNA_seq.fastq- Convert unmapped reads (using bamToFastq from bedtools)
bamToFastq -i tophat/unmapped.bam -fq tophat/unmapped.fastq- Unique mapping with TopHat-Fusion
tophat2 -o tophat_fusion -p 15 --fusion-search --keep-fasta-order --bowtie1 --no-coverage-search hg19_bowtie1_index tophat/unmapped.fastqTo detect fusion junctions with STAR, --chimSegmentMin should be set to a positive value.
Example 1 (single reads):
STAR --chimSegmentMin 10 --runThreadN 10 --genomeDir hg19_STAR_index --readFilesIn RNA_seq.fastqExample 2 (paired-end reads):
STAR --chimSegmentMin 10 --runThreadN 10 --genomeDir hg19_STAR_index --readFilesIn read_1.fastq read_2.fastqFor more details about STAR, please refer to STAR manual.
pip install CIRCexplorerCIRCexplorer is available as conda package with:
conda install circexplorer --channel biocondaIf you have access to a Galaxy instance, CIRCexplorer is also available from the Galaxy Tool Shed.
1 Download CIRCexplorer
git clone https://github.com/YangLab/CIRCexplorer.git
cd CIRCexplorer2 Install required packages
pip install -r requirements.txt3 Install CIRCexplorer
python setup.py install
CIRCexplorer.py 1.1.10 -- circular RNA analysis toolkits.
Usage: CIRCexplorer.py [options]
Options:
-h --help Show this screen.
--version Show version.
-f FUSION --fusion=FUSION TopHat-Fusion fusion BAM file. (used in TopHat-Fusion mapping)
-j JUNC --junc=JUNC STAR Chimeric junction file. (used in STAR mapping)
-g GENOME --genome=GENOME Genome FASTA file.
-r REF --ref=REF Gene annotation.
-o PREFIX --output=PREFIX Output prefix [default: CIRCexplorer].
--tmp Keep temporary files.
--no-fix No-fix mode (useful for species with poor gene annotations)CIRCexplorer.py -f tophat_fusion/accepted_hits.bam -g hg19.fa -r ref.txt- convert
Chimeric.out.junctiontofusion_junction.txt(star_parse.pywas modified from STAR filterCirc.awk)
star_parse.py Chimeric.out.junction fusion_junction.txt- parse
fusion_junction.txt
CIRCexplorer.py -j fusion_junction.txt -g hg19.fa -r ref.txt- ref.txt is in the format (Gene Predictions and RefSeq Genes with Gene Names) below (see details in the example file)
| Field | Description |
|---|---|
| geneName | Name of gene |
| isoformName | Name of isoform |
| chrom | Reference sequence |
| strand | + or - for strand |
| txStart | Transcription start position |
| txEnd | Transcription end position |
| cdsStart | Coding region start |
| cdsEnd | Coding region end |
| exonCount | Number of exons |
| exonStarts | Exon start positions |
| exonEnds | Exon end positions |
-
hg19.fa is genome sequence in FASTA format.
-
You could use fetch_ucsc.py script to download relevant ref.txt (Known Genes, RefSeq or Ensembl) and the genome fasta file for hg19, hg38 or mm10 from UCSC.
fetch_ucsc.py hg19/hg38/mm10 ref/kg/ens/fa outExample (download hg19 RefSeq gene annotation file):
fetch_ucsc.py hg19 ref ref.txtSee details in the example file
| Field | Description |
|---|---|
| chrom | Chromosome |
| start | Start of junction |
| end | End of junction |
| name | Circular RNA/Junction reads |
| score | Flag to indicate realignment of fusion junctions |
| strand | + or - for strand |
| thickStart | No meaning |
| thickEnd | No meaning |
| itemRgb | 0,0,0 |
| exonCount | Number of exons |
| exonSizes | Exon sizes |
| exonOffsets | Exon offsets |
| readNumber | Number of junction reads |
| circType | 'Yes' for ciRNA, and 'No' for circRNA (before 1.1.0); 'circRNA' or 'ciRNA' (after 1.1.1) |
| geneName | Name of gene |
| isoformName | Name of isoform |
| exonIndex/intronIndex | Index (start from 1) of exon (for circRNA) or intron (for ciRNA) in given isoform (newly added in 1.1.6) |
| flankIntron | Left intron/Right intron |
Note: The first 12 columns are in BED12 format.
Copyright (C) 2014-2017 YangLab. See the LICENSE file for license rights and limitations (MIT).