Tutorial
BiERapp is an interactive web application for assisting in gene prioritization in whole exome sequencing (WES) experiments.
This section explains how to use main tasks for BiERapp:
- On the right top of home page, there are two buttons Login and Sign up, and a help option that contains: documentation and tutorial.

- After logging in, other options can be visualized on the top of main page.

- To create a new account in BiERapp, you only have to click sign in. It will appear a new window where you can select new account and fill in some fields. It is free. Once your account is created you can log in into a private session. All the jobs you run and all the panels you create will be saved there and will appear in the next session.
- We'll see the different functionalities of the tool.
From My Data or from Study Browser you can:
- Create a Project
- Create a Study
- Upload Files: The input is a VCF file: individual VCF file for only one sample or multisample VCF file for several samples (families or group of cases /controls).
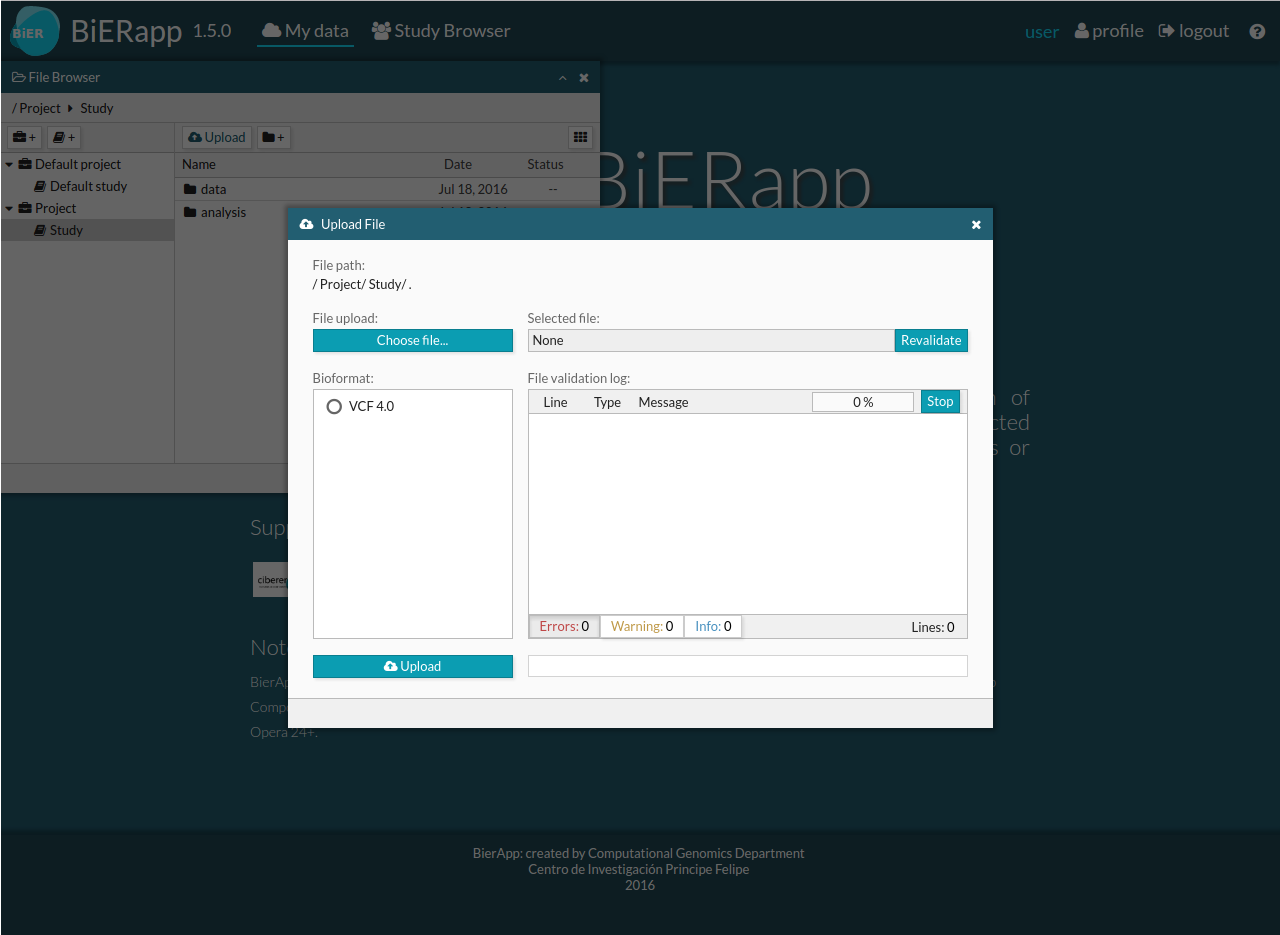
You must click in Upload button and a new window will open. You can choose a VCF file and wait to finish the validation. If there is no errors in your file,then you can click in Upload and the VCF will load and begin automatic indexing. If there are one or more errors can not load the file, you must correct the errors shown in errors tab. If there is more than hundred errors, validation is stopped automatically. There is also a stop button and a revalidate button.
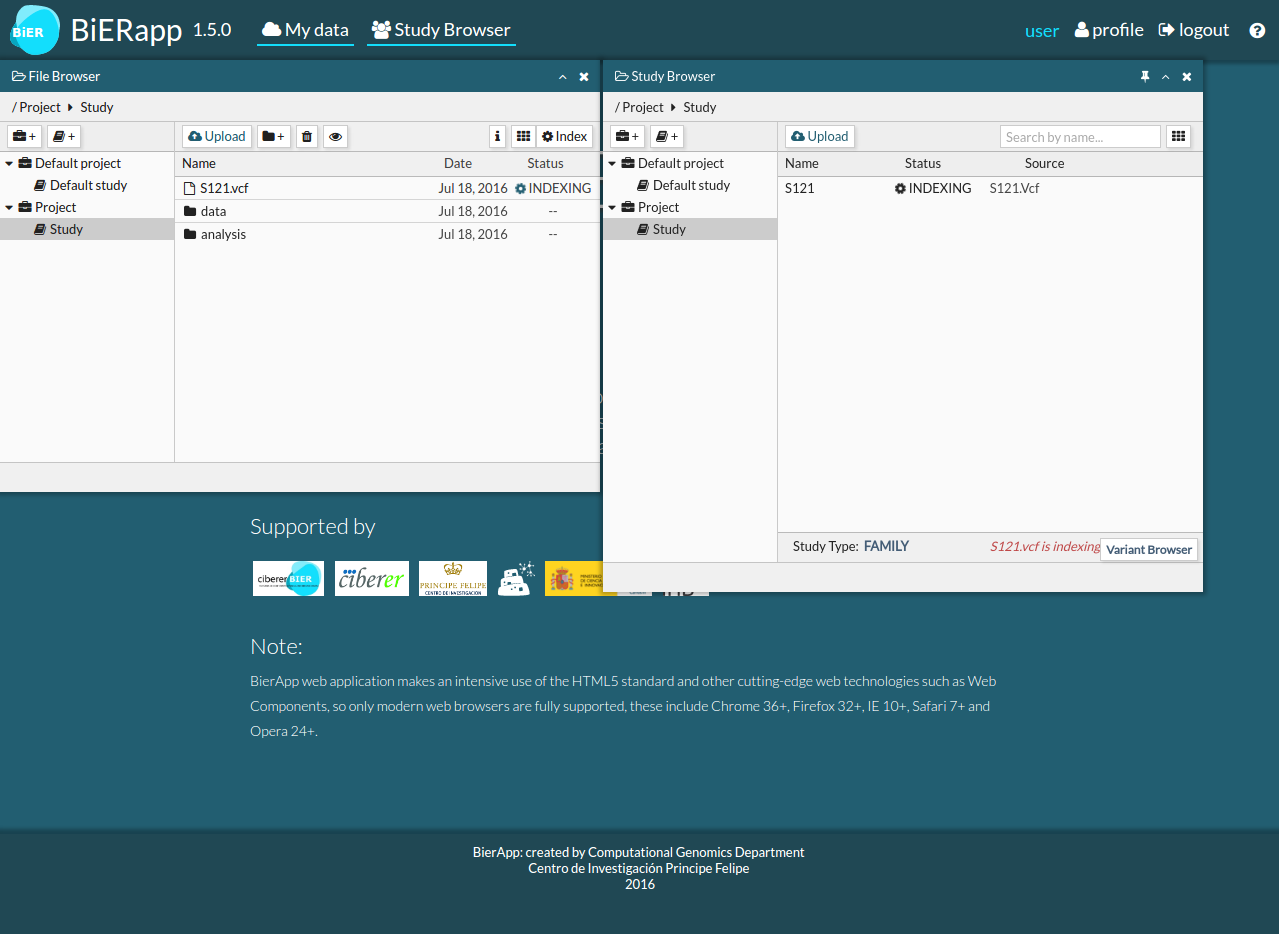
- After uploading VCF file, BiERapp detects variants for all samples.
- You can select one or more samples in READY status to see them in Variant Browser.
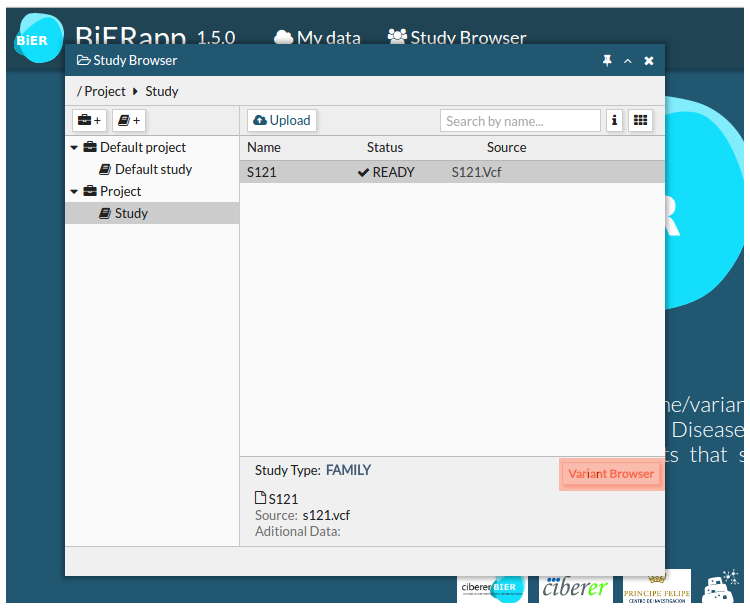
- Initial results show the whole set of variants. This is a general first view of detected variants. Next step for each user is defining his own strategy of filtering according to his interests.
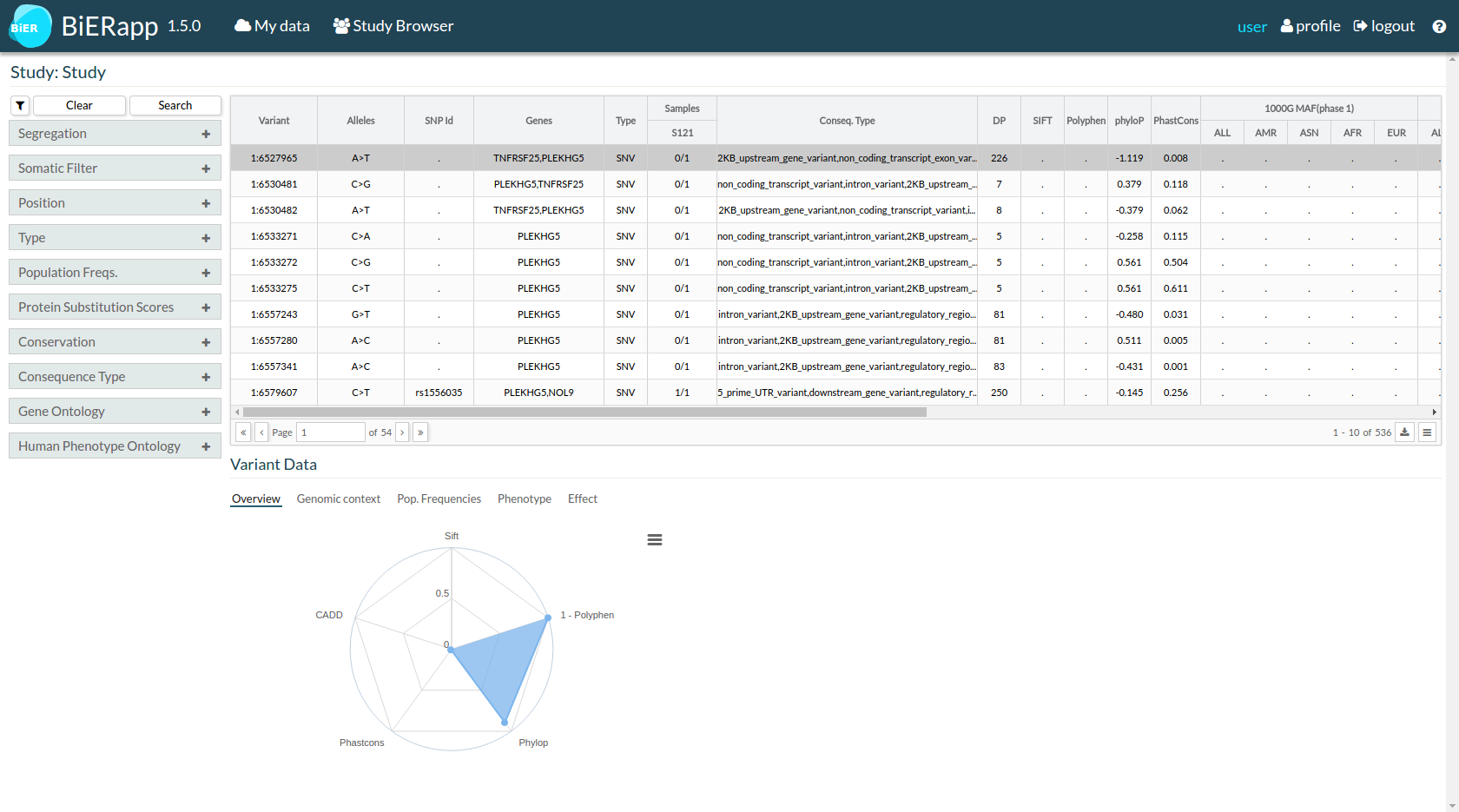
Clicking drop down buttons, it is possible order this information for any of these variables.
There are several filters to define the best strategy of variant selection. It is possible to choose one or several filtering options at the same time:
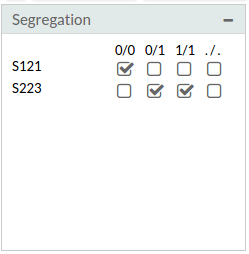
- Segregation: This intuitive filter that allows reproducing any familiar pedigree with any inheritance model. Also case-control or sporadic de novo mutational diseases can be analysed in this framework. BiERapp also manages efficiently missing values (by default BiERapp shows variants with 0 missing values for genotypes, but you can modify this parameter). We only have to select the possible genotypes for each sample.
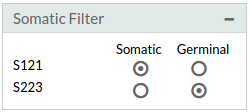
- Somatic: You can choose Somatic or Germinal samples and search intersection.
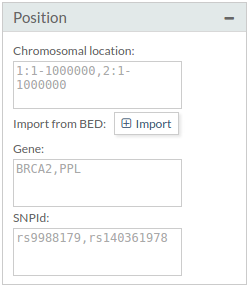
- Position:
- Region: Several possibilities to define a region:
- Only one chromosomal region: 1:1-10000000
- Several regions (separated by comma): 1:1-10000000, 2:1-10000000
- Some chromosomes(separated by comma): 4,5,8 - Gene: Only variants for a group of genes. Example: BRCA2,PPL (separated by comma)
- SNPId: Example: rs9988179,rs140361978 (separated by comma)
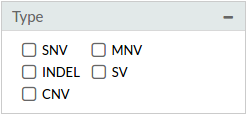
- Type: SNV, INDEL, CNV, MNV, SV
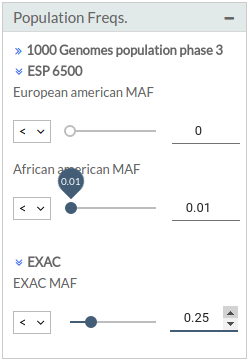
- Population frequencies: This filter is based on known population frequencies, including several subpopulations:
- 1000 Genomes Phase 3: 1000 Genomes
- ESP 6500: Exome Variant Server
- ExAC: [Exome Aggregation Consortium] (http://exac.broadinstitute.org/)
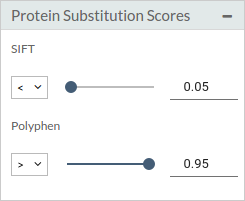
- Protein substitution scores:
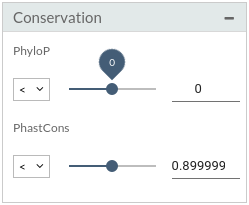
- Conservation: Set pathogenicity thresholds.
- PhastCons is a program for identifying evolutionarily conserved elements in a multiple alignment, given a phylogenetic tree.
- PhyloP scores measure evolutionary conservation at individual alignment sites
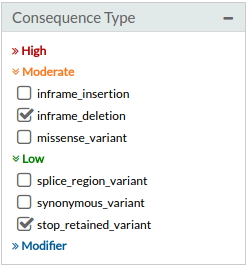
- Consequence types: Effect. Selecting one or multiple consequence type. It is possible uncheck or selecting several effects at the same time. The terms are sorted by the [IMPACT] (http://www.ensembl.org/Help/Glossary?id=535) rating of the severity of the variant consequence (high, moderate, low or modifier).
- Gene ontology: [GO] (http://geneontology.org/)
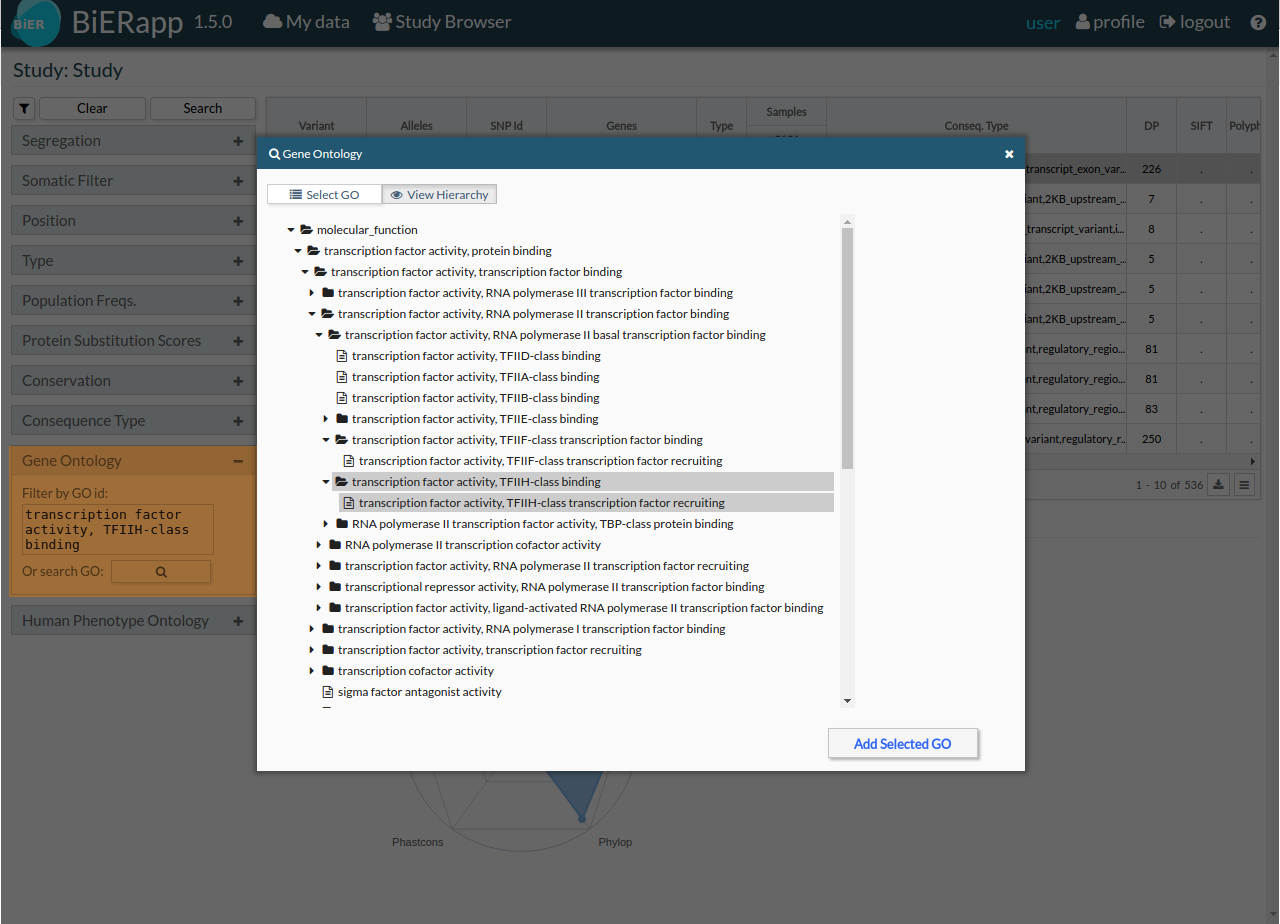
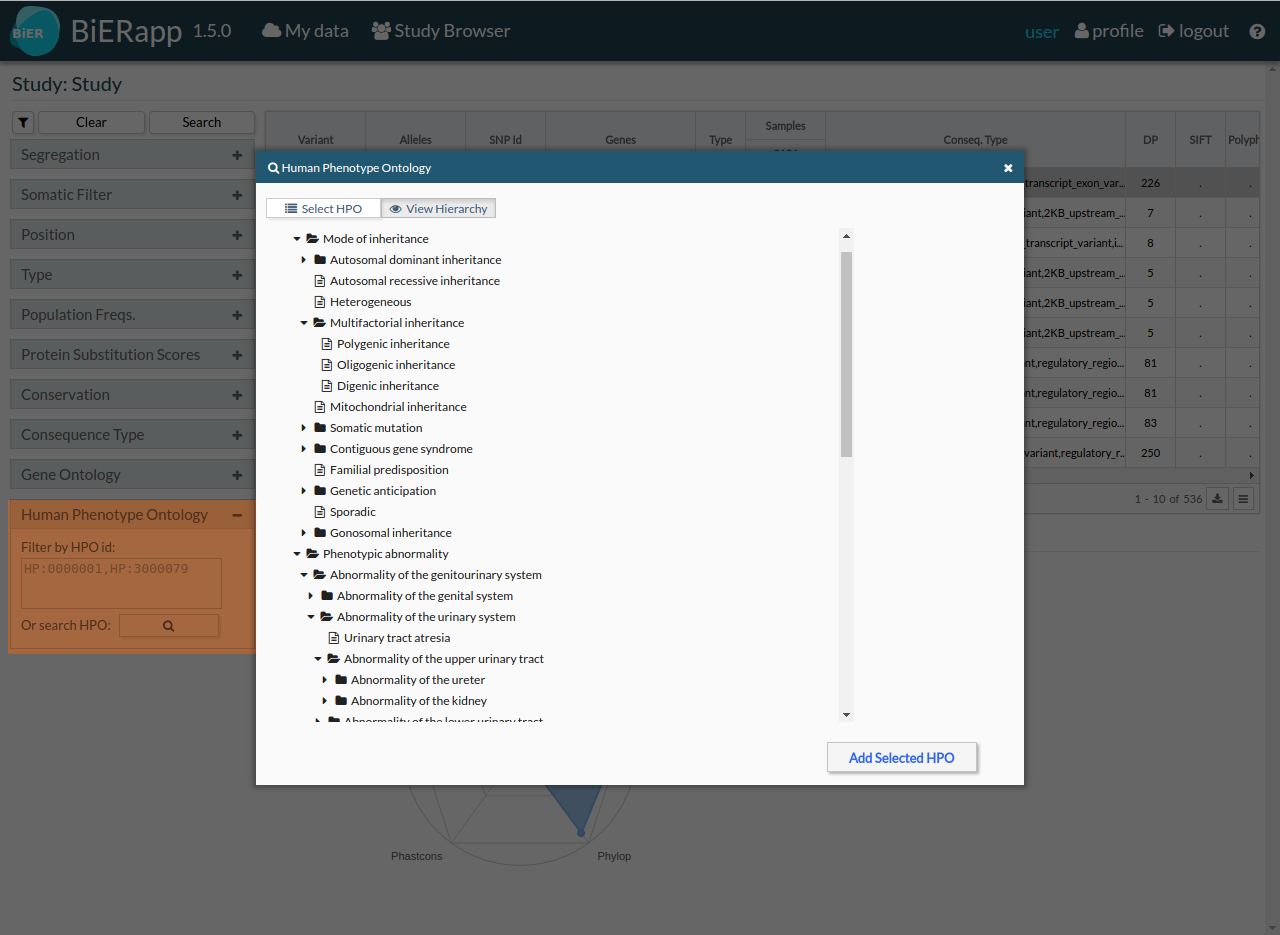
- Human Phenotype Ontology: [HPO] (http://human-phenotype-ontology.github.io/)
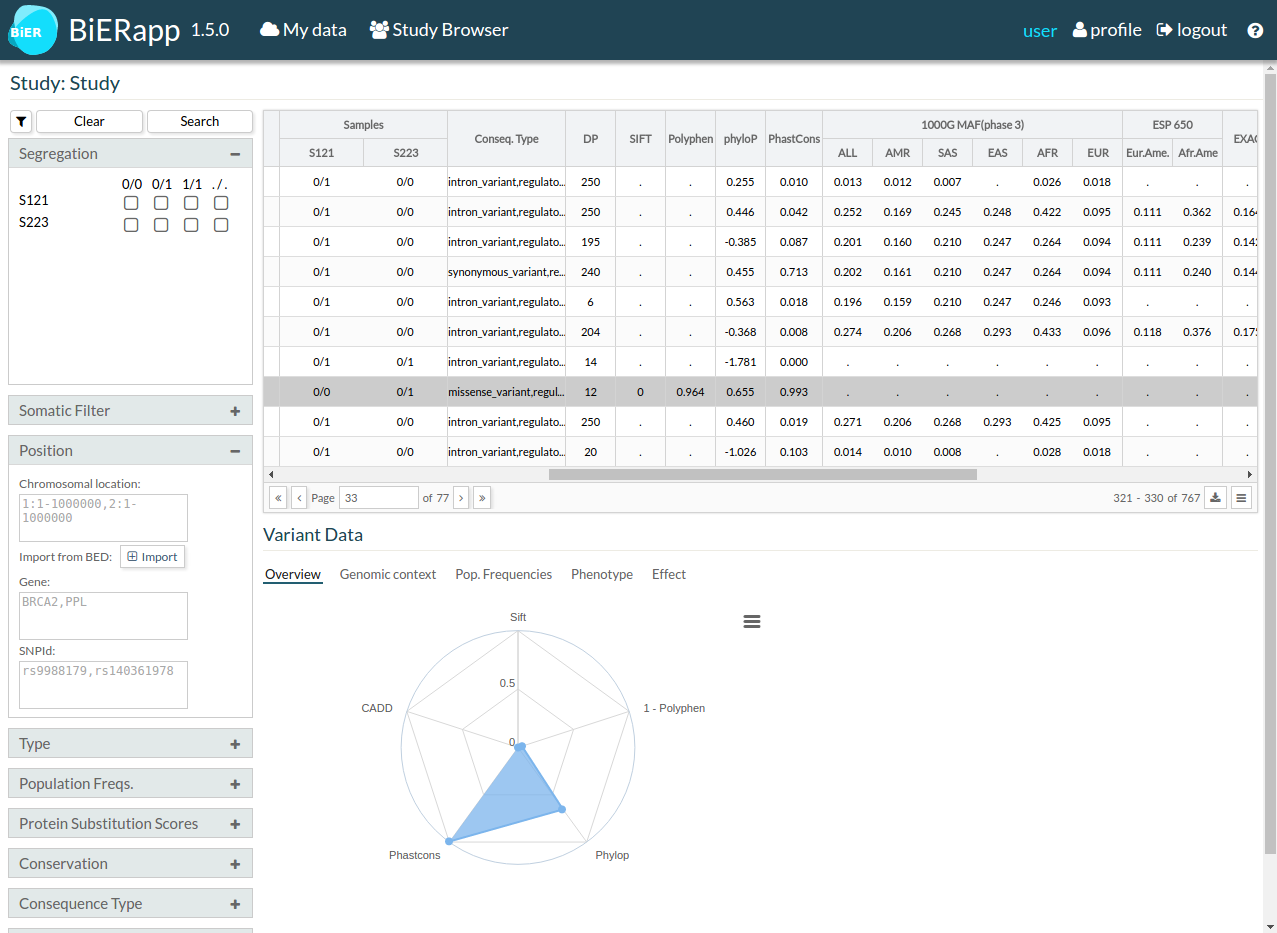
Under the table of variants you can find more information about a selected variant.
- First tab Overview shows a graph about protein substitution scores and conservation scores.
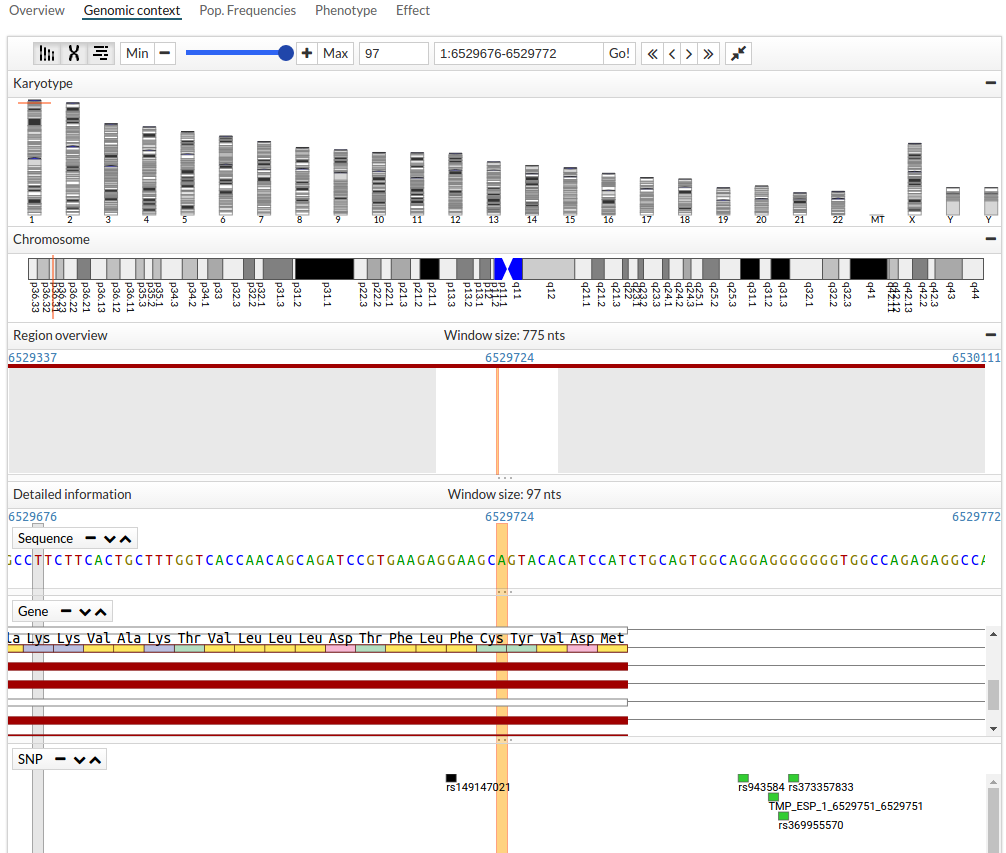
- Second tab Genomic context shows the Genome Viewer.
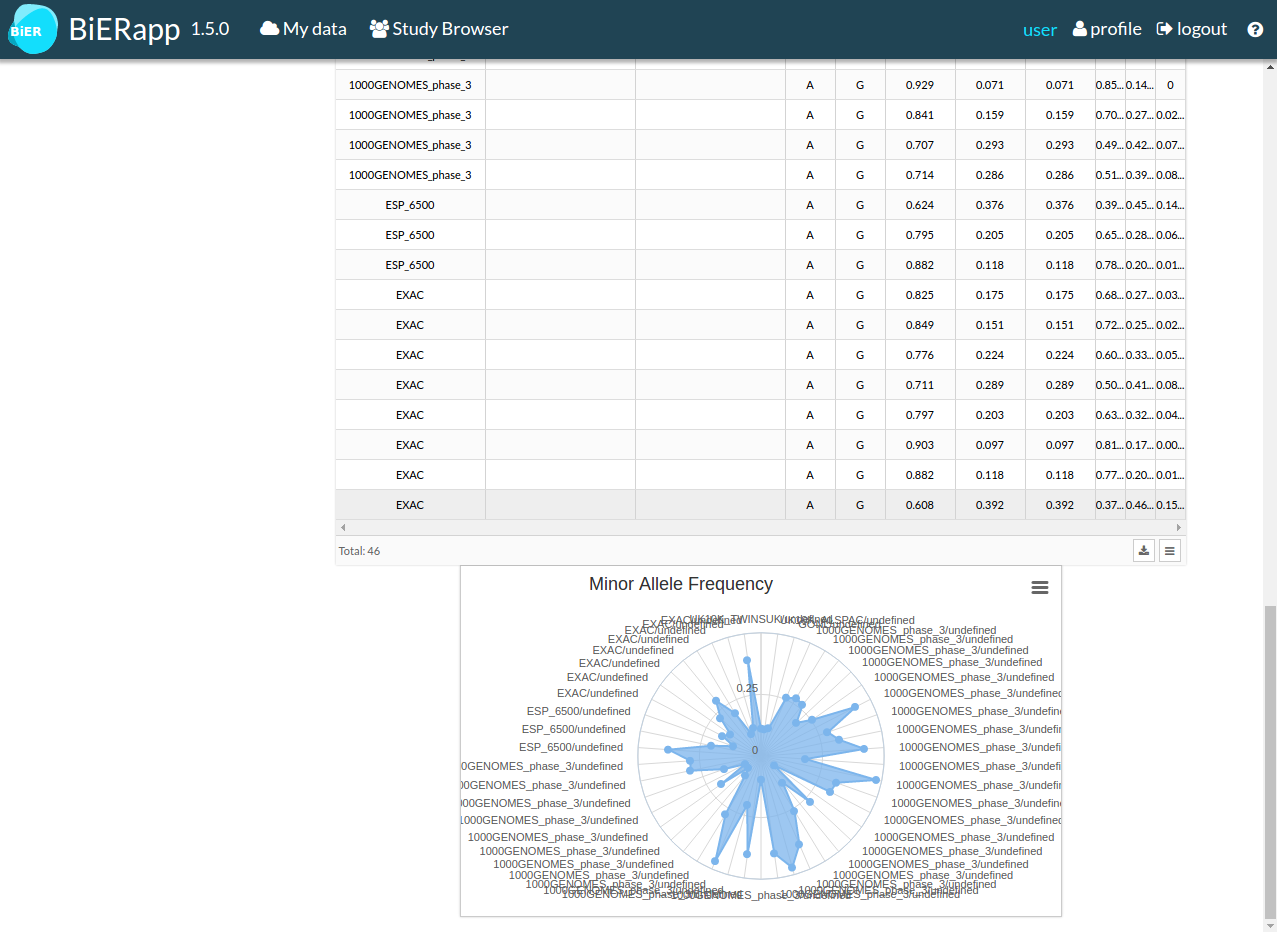
- Third tab Pop.Frequencies shows all frequencies asociated to the selected variant, in table format and in graph format.
- Fourth tab shows the Effect information for each variant:
To report an error or suggestion, please contact us at: babelomics@cipf.es