-
Notifications
You must be signed in to change notification settings - Fork 12
CNV seq
Here we show an implementation of CNV-seq. An R script using the GenomicRanges infrastructure for binning and counting, rather than the perl script in CNV-seq, is available in the script RCNV_seq.R.
Step - 1 This includes generating best-hit location files for each mapped sequence read. The authors provide a perl script for BLAT psl file and SOLiD maching pipeline. For BAM files, they suggest to extract locations using the following command
~/copynumber$ samtools view -F 4 tumorA.chr4.bam |perl -lane 'print "$F[2]\t$F[3]"' >tumor.hits
~/copynumber$ samtools view -F 4 normalA.chr4.bam |perl -lane 'print "$F[2]\t$F[3]"' >normal.hits
This command says to select reads with flag '4' (the read is mapped) and to print out the 'rname' and 'pos' of their alignment.
Step - 2 cnv-seq.pl is used to calculate sliding window size, to count number of mapped hits in each window, and to call cnv R package to calculate log2 ratios and annotate CNV
perl cnv-seq.pl --test tumor.hits --ref normal.hits --genome human
two output files are produced. They can be found under "result files"
tumor.hits-vs-normal.hits.window-10000.minw-4.cnv
tumor.hits-vs-normal.hits.window-10000.minw-4.count
Step - 3
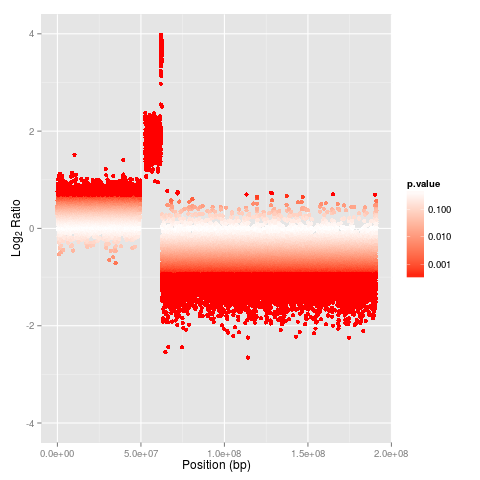
One can visualize the cnv inside R using the following code snippet
the plot can be found under "image" folder
![] (https://raw.github.com/Bioconductor/copy-number-analysis/master/image/cnv-seq-plot.png)
{kind=link}
The R code for visualizing the plot can be found at : CNV-seq.R